[Previous: Chapter 4: MFO]
[Next: Chapter 6: General Discussion]
Rule-based mediation for the semantic integration of systems biology data
Introduction
The creation of accurate quantitative SBML models is a time-intensive manual process. Modellers need to know and understand both the systems they are modelling and the intricacies of the SBML format. However, the amount of relevant data for even a relatively small and well-scoped model can be overwhelming. Systems biology model annotation in particular can be time-consuming; the data required to perform the annotation is represented in many different formats and present in many different locations. The modeller may not even be aware of all suitable resources.
There is a need for computational approaches that automate the integration of multiple sources to enable the model annotation process. Ideally, the retrieval and integration of biological knowledge for model annotation should be performed quickly, precisely, and with a minimum of manual effort. If information is added manually, it is very difficult for the modeller to annotate exhaustively. Current systems biology model annotation tools rely mainly on syntactic data integration methods such as query translation. A review of data integration methodologies in the life sciences is available in Section 1.6, and the Saint syntactic integration system for model annotation is described in detail in Chapter 3.
While large amounts of data can be gathered and queried with syntactic integration, the integration process is limited to resolving formatting differences without attempting to address semantic heterogeneity, which occurs when a concept in one data source is matched to a concept in another data source which is not exactly equivalent. Semantic data integration methods can be used to resolve differences in the meaning of the data. This chapter describes rule-based mediation, a novel methodology for semantic data integration. While rule-based mediation can be used for the integration of any domain of interest, in this instance it has been applied to systems biology model annotation. Off-the-shelf Semantic Web technologies have been used to provide novel annotation and interactions for systems biology models. Existing tools and technology provide a framework around which the system is built, reducing development time and increasing usability.
There are three main parts to rule-based mediation: the source ontologies for describing the disparate data formats, the mapping rules for linking the source ontologies to a biologically-relevant core, and the core ontology for describing and manipulating semantically-homogeneous data (see Figure 1). In rule-based mediation, the data is materialised in the core ontology by using a set of rules to translate the data from the source ontologies to the core ontology, avoiding the need for complex query translation algorithms.
Rule-based mediation has a number of defining features:
- the data from syntactically and semantically different data sources are materialised within a core ontology for easy access and stable querying;
- the core ontology is a semantically-meaningful model of the biological domain of interest; and
- implementation can be performed using off-the-shelf tools and mapping languages.
Integrating resources in this way accommodates multiple formats with different semantics and provides richly-modelled biological knowledge suitable for annotation of systems biology models. This work establishes the feasibility of rule-based mediation as part of an automated systems biology model annotation system.
Section 1 describes the type of integration performed in rule-based mediation as compared with existing data integration methodologies. The first step in rule-based mediation is the syntactic conversion of heterogeneous data sources into ontologies, as detailed in Section 2. These source ontologies are then aligned to a small core ontology by applying a rule base (Section 3). This core ontology is a model of the biological domain of interest which can be queried and reasoned over (Section 4). As demonstrated in Section 5 with two use cases, selected results from the core domain ontology can then be applied to systems biology models as a variety of types of biological annotation. Section 6 describes possible future directions and applications for rule-based mediation, and Section 7 provides a final discussion and summary of the methodology.
Availability
The rule-based mediation website is http://cisban-silico.cs.ncl.ac.uk/RBM/, and the ontologies and software toolset is available from Subversion (Note: http://metagenome.ncl.ac.uk/subversion/rule-based-mediation/telomere/trunk). The ontologies stored within the rule-based mediation project are licensed by Allyson Lister and Newcastle University under a Creative Commons Attribution 2.0 UK: England & Wales License (Note: http://creativecommons.org/licenses/by/2.0/uk/). The rule-based mediation software toolset is covered under the LGPL (Note: http://www.gnu.org/copyleft/lesser.html). For more information, see LICENSE.txt in the Subversion project.
The SWRL rules described in this chapter can be run programmatically or via the Protégé 3.4 editor (Note: http://protege.stanford.edu). Programmatic access requires the rule-based mediation software toolset, which in turn requires the OWL API. The SWRLTab plugin within Protégé 3.4 requires the installation of the Jess Rule Engine (Note: http://www.jessrules.com/) for Java. All mapping rules are available for viewing within swrl-mappings.owl in the Subversion distribution. Instructions for viewing the telomere ontology, the source ontologies, the SWRL mappings, SQWRL queries, and the SBML model before and after annotation in these use cases are available in the Subversion repository (Note: http://metagenome.ncl.ac.uk/subversion/rule-based-mediation/telomere/trunk/INSTALL.txt).
1 Design decisions
This section describes the design decisions made for rule-based mediation with respect to existing methodologies. An overview of this integration methodology is summarised in Figure 2 in the context of SBML model annotation. First, information is syntactically converted into OWL or retrieved, correctly formatted, in an pre-existing source ontology. Second, the information is mapped into a core ontology. Third, the core ontology is queried to answer specific biological questions. Finally, the new information is sent back to an external format, in this case SBML, via mappings from the core ontology to MFO. From a biological perspective, rule-based mediation produces an integrated view of information useful for modelling.
1.1 Syntactic or semantic integration
The majority of systems biology model annotation tools such as those described in Section 1.3 rely on syntactic data integration methods. Taverna workflows can act as query translation services to pull annotation into models from data sources [1]. SemanticSBML [2] provides MIRIAM annotations, but does not create any other model elements. LibAnnotationSBML connects to and serves data from a large number of Web services useful for model annotation but has only a minimal user interface [3]. BioNetGen combines its own rules of molecular interactions with BioPAX-compliant data and the Virtual Cell editor to produce SBML [4]. However, BioNetGen requires some level of manual intervention and neither adds MIRIAM annotations nor integrates disparate data formats, instead accepting only BioPAX-formatted data. Chapter 3 describes Saint, which uses query translation and an intuitive user interface to pull information from multiple Web services and syntactically integrate the results, which the user can then select and apply to a pre-existing model [5].
However, while large amounts of data can be gathered and queried in this manner, the syntactic integration process is limited to resolving formatting differences without attempting to address semantic heterogeneity. While existing syntactic tools for model annotation are suitable for many tasks, semantic data integration methods can resolve differences in the meaning of the data and theoretically provide a more useful level of integration. A number of semantic integration approaches have been applied to many different research areas [6, Section 9], and are beginning to be used in the life sciences (see also Sections 1.6 and 1.5).
Formal ontologies are useful data integration tools, as they make the semantics of terms and relations explicit [7]. They also make knowledge accessible to both humans and machines, and provide access to automated reasoners. By modelling source data and the core domain of interest as ontologies, semantically-meaningful mapping can be used to link them. Such mappings are not possible with hard-coded syntactic conversions. The use of ontologies provides a rich integration environment, but can be costly with regards to the time taken to analyse and reason over the data. The primary cost is the scalability of ontology-based integration methods. As ontologies grow in size and complexity, the reasoning time rapidly increases.
1.2 Mapping type for the integration interface
As described in Section 1.6, neither information linkage nor direct mapping meets the needs of this project. Information linkage is not true integration, and direct mapping is not suitable for situations where more than two resources need to be integrated. EKoM [8] and SBPAX [9] are two semantic direct mapping approaches discussed in Section 1.6 which most closely match the goals of rule-based mediation. However, modification of either of these approaches is not feasible, as they were both developed to integrate only two resources.
The integrated data in EKoM [8] is presented in RDF rather than OWL. The high scalability of RDF compared with OWL comes at the expense of expressivity. Although large amounts of data can be efficiently stored and queried with RDF, the RDF query language SPARQL cannot make use of OWL, thus limiting the possible query space. There are other limitations to the EKoM method: the addition of new data in different formats would require the modification of the main ontology, and perhaps even the stored queries used to retrieve information from it; and there is no mention of any ability to export results using one of the import formats, as is possible with rule-based mediation.
SBPAX, originally developed to integrate SBML and BioPAX, seems to provide much of the required annotation functionality for systems biology models. Extension of this method to more than two ontologies by expansion of the role of the bridging ontology initially sounds feasible. However, SBPAX was not created to be a generic methodology and the latest release (SBPAX3) has a different purpose from previously published versions (Note: http://sbpax.org/sbpax3/index.html). Instead of being created for purely integrative purposes, SBPAX3 has moved away from ontology mapping and is instead a proposed extension to BioPAX Level 4 to incorporate quantitative information into that standard. While SBPAX3 does allow closer alignment between the SBML XML format and the BioPAX OWL format, its primary purpose has moved from data integration to extension of BioPAX functionality, making this methodology highly unlikely to incorporate additional data sources in future.
Because of the limitations with non-mediator methods, a mediator methodology has been used for rule-based mediation. Within the three mediator types described in Section , multiple mediator mapping was rejected due to its complexity; this mapping subtype requires that the users, whether machine or human, be aware of the multiple data models. As a result semantic solutions using this approach, such as linked data or RDF triple stores, are not integrated enough to be useful for this project.
A single unified view makes examination, querying and ultimately viewing the integrated dataset easier. This leaves either single mediator mapping or hybrid mediator mapping. Hybrid mediator mapping was rejected as it requires that each source ontology be aware of the global ontology, thus immediately making it impossible to utilise ontologies with no knowledge of rule-based mediation such as pre-existing community ontologies. Therefore single mediator mapping, which has the benefit of being a straightforward mapping method with minimal pre-requisites, was chosen.
There are three subtypes within single mediator mapping: global-as-view, local-as-view, and hybrid approaches. While the global-as-view approach makes the passing of queries to the underlying data sources simple, it has other restrictions which make it unsuitable for model annotation. For instance, defining the global ontology merely as a model of a set of data sources creates an ontology which is brittle with respect to the addition of new data sources and new formats. However, choosing local-as-view can also be problematic, as query complexity increases quickly with local-as-view approaches, and data sources need to be forced into the structure of the global ontology [10].
Both methods are limited in their ability to describe the nuances of the biological concepts being studied. When one layer (either the global or source ontologies) is a view of the other, conflation of format and biological domain concepts into a single model can occur. For global-as-view, the source ontologies are intended to model the data sources, information which is of no use in a global ontology intended to be a model of a biological domain of interest, as in rule-based mediation. For local-as-view, the source ontologies are merely views over a global ontology, restricting the description of the syntax of the sources.
Therefore, hybrid approaches which do not force one layer to be the view of another provide a more flexible solution. The BGLaV [10] project describes a hybrid mapping method where the global ontology is modelled completely separately from the source ontologies. However, its overall methodology depends upon complex query translation algorithms to pull information from the underlying sources [10]. Therefore rule-based mediation uses this hybrid mapping type but creates a new methodology using existing Semantic Web mapping strategies which does not have the query complexity of BGLaV.
1.3 The integration interface: warehousing or federation
The final refinement to the methodology is the integration interface itself, and the decision as to whether a warehousing or federated approach is appropriate for rule-based mediation. While warehousing methods (described fully in Section 1.6) are powerful and quick, they are not scalable and can create large resources which current ontological reasoning tools struggle to process. The Neurocommons integration methodology, which uses data encapsulation via multiple mediation mapping, partially solves these scalability issues [11]. While OWL is used to re-format the original data sources, Neurocommons minimises the reasoning cost by using plain RDF as the format for the main data store, which does not get reasoned over. Further, while Neurocommons makes use of Semantic Web technologies, they integrate at the level of the database entry itself [12] and do not try to model the biological concepts referenced by those entries. As the focus of rule-based mediation is on providing modellers with a method of targeted annotation of specific models, such a large triple store is not required, and a methodology generating a smaller, more semantically-rich dataset amenable to reasoning is required.
Another ontology-based model comparison system has recently been developed by Schulz and colleagues to integrate the semantic MIRIAM annotations in SBML across different models in the BioModels database [13]. While this method is useful for finding semantically similar entities in SBML, it is not intended as a large-scale, multiple data source integration methodology.
As described in Chapter 4, SBML Harvester allows the loading and querying of SBML models in OWL. Both rule-based mediation and SBML Harvester apply automated reasoning to enrich the knowledge available to an SBML model. SBML Harvester also makes use of an upper-level ontology that integrates in silico and in vivo information and includes a number of community ontologies [14]. However, the conflation of both structural and biological entities in a single ontology could make it more difficult to clearly isolate syntactic from semantic heterogeneity. Additionally, Hoehndorf and colleagues have stated that the existing biomedical ontologies used within the system often have a weak formalisation which limits the capabilities of SBML Harvester for verification and knowledge extraction [14]. The authors have expressed a desire to see if rule-based mediation and SBML Harvester can be combined in future [14].
Data federation (see Section 1.6) provides its own challenges. Federated approaches require constant calls to remote Web sources to retrieve the requested subset of information and are not guaranteed to be able to access source data. Older federated integration techniques such as TAMBIS use only a single ontology, mapping both the semantics and syntax of the underlying data sources into that ontology [15]. TAMBIS also assumes only one data source per format type. In contrast, rule-based mediation separates the tasks of resolving syntactic and semantic heterogeneity through the use of source ontologies as well as a core ontology.
Query translation systems such as ComparaGRID suffer from inefficiency during data retrieval. With local-as-view methods such as OntoFusion, input data can only be modelled as a view over the global ontology. As the global ontology is independent of the data sources, it may not have the expressivity to model all of the information in every format of interest. In PISCEL [16], class and query rewriting were used to pull information from each data source at the time of the query. However, in rule-based mediation the core ontology is a tightly-scoped ontology which benefits from having all of the relevant information stored directly within it, thus obviating the need for a query rewriting step.
Large-scale ontology alignment has very recently been performed using a variety of OBO Foundry ontologies by aligning each ontology to a upper-level philosophical ontology [7]. While this method has found numerous modelling errors, inconsistencies, and contradictions in the aligned ontologies, integration is limited to the knowledge within the ontologies themselves and is not attempted for any instance-level data or biological databases. It provides valuable error checking across ontologies which are intended to be orthogonal according to OBO Foundry principles (Note: http://www.obofoundry.org/wiki/index.php/FP_005_delineated_content), but is a methodology tailored to ontologies without instance data, making it unsuitable for this thesis.
The integrated data in rule-based mediation needs stability to ensure effective querying, but also needs to be lightweight enough that reasoning can be performed. As such, rule-based mediation has aspects of both warehousing and federation. In rule-based mediation, instance data is stored in the core ontology for as long as a user wishes, creating a warehouse of integrated data. However, the user selects only a subset of each data source to be loaded into the core ontology. The query results are then filtered through the source ontologies and mapped to the core. This query step can be performed repeatedly, adding or removing information from the global ontology as required, thus providing a federated quality to the methodology. Because the core ontology in rule-based mediation is a tightly-scoped ontology modelling a specific biological domain of interest, only the information required for a particular use-case needs to be added.
1.4 Ontology mapping rules
SWRL [17] is a Semantic Web rule language that extends OWL with the addition of a new if-then conditional and a number of convenience methods called built-ins [18]. The DL-safe version of SWRL is used within rule-based mediation to create mappings, also called rules, from one entity to another. SWRL has mature libraries and is interoperable with the existing OWL API. Although it is possible to hard code rules using a library such as the OWL API, SWRL rules allow the logical separation of the knowledgebase and the rulebase from any lower-level code, make knowledge shareable across ontologies, and make the knowledge easier to maintain [19].
SWRL rules are divided into a antecedent and a consequent. In the simple case below, both the antecedent (classA(?X)) and the consequent (classB(?X)) are classes, though SWRL is often used for more complex assertions involving properties, classes and even instances:
classA(?X) → classB(?X)
In SWRL, whenever the antecedent of the rule is true, the conditions in the consequent must also hold. The above mapping in plain English is “if X is a member of classA, then X is a member of classB”. With this mapping, all instances ?X of classA are also asserted as members of classB. classA and classB could be from the same or different ontologies. SWRL rules provide only instance-to-instance mapping, which results in the assignment of instance data to membership in another class. No classes or properties are ever modified by SWRL.
SWRL is not the only way to create rules in OWL. Two or more OWL ontologies can be linked together with a number of methods. In ontology alignments, classes can be marked as equivalent or joined together via other, more complex, axioms (see Section 1.6 for a discussion of ontology alignment and merging). This method does not require any languages other than OWL itself, and is easily visible within the asserted hierarchy. However, each aligned ontology must be imported into the same file; imports can cause problems if the ontologies are large or numerous, and it is impractical if only part of the ontology needs to be aligned. DL rules [20] are pure OWL and are a fragment of SWRL. However, the use of complete rule languages such as SWRL is often easier than OWL rules and can sometimes be a requirement for expressing particular mappings (Note: http://answers.semanticWeb .com/questions/1354/why-do-we-need- swrl-and-rif-in-an-owl2-world, http://answers.semanticWeb .com/questions/2329/rifspin-needed-or-not-as-when-owl2-seems-sufficient).
Although multiple implementations for ontology mapping exist, ultimately only the SWRLTab [21] plugin for Protégé 3.4 and the OWL API were used within rule-based mediation. SWRLTab is a Protégé 3.4 plugin which can perform ontology mapping as well as query relational data sources or XML, and was used within rule-based mediation to create and execute SWRL rules. SWRL is used for the mapping rules and SQWRL [22] for querying those rules. While SWRL rules are written using OWL and are therefore easy to understand, there are drawbacks to this implementation. SWRLTab requires that each additional ontology to be mapped must be imported directly into the main ontology. Therefore when mapping many source ontologies to a single core ontology, the amount of imports could become unwieldy. Further, importing via the normal OWL import mechanism means that multiple dissimilar ontologies are combined in one logical area. However, once the SWRL rules have been run, a cleaner version of the final ontology—without the source ontologies or the rule file—may be viewed if desired.
Other ontology mapping applications were investigated and ultimately deemed inappropriate for rule-based mediation. Snoggle (Note: http://snoggle.semWeb central.org/) is a graphical, SWRL-based ontology tool for aiding alignment of OWL ontologies. Ontology alignment is mainly used for merging two ontologies and as such is not ideally suited to rule-based mediation, which retains the independence of each ontology. Similarly, Chimera (Note: http://www-ksl.stanford.edu/software/chimaera/) is intended for alignment and manipulation, but not mapping. COMA++ (Note: http://dbs.uni-leipzig.de/Research/coma.html) currently only supports OWL-Lite. While capable of limited OWL functionality, COBrA [23] is primarily for OBO files, specifically GO. Falconer and colleagues provide an overview of a number of other ontology mapping tools [24].
Closer to the requirements of this rule-based mediation are PROMPT [6] and Fluxion (Note: http://bioinf.ncl.ac.uk/comparagrid/). PROMPT is a Protégé plugin which performs automatic mapping and allows user-provided manual mappings. It is a mature application with the ability to map classes, relations, and instances. However, PROMPT could not provide any automated mappings between the BioGRID syntactic ontology and the telomere ontology. Fluxion is a standalone ontology mapping tool which has been used within the ComparaGrid system, and is a service-oriented architecture for federated semantic data integration which uses translation rules to map between the source ontologies and the core ontology (Note: http://bioinf.ncl.ac.uk/comparagrid/). However, Fluxion is not currently in development and cannot be easily integrated with the overall structure of rule-based mediation.
Querying an ontology
The simplest method of ontology querying is the creation of defined classes which function as queries over the instance data, as with Luciano and colleagues [25]. Alternatively, SPARQL queries can be constructed for ontologies which have an underlying RDF. However, both methods have their limitations. Defined classes created as queries are difficult to add on-the-fly, and have similar limitations the use of plain OWL does for the creation of rules. SPARQL is only compatible with RDF-formatted ontologies. SQWRL is a query language based on SWRL which is used for querying OWL ontologies and formatting the query response. Unlike SWRL, SQWRL does not modify the underlying ontology. Therefore, SQWRL queries can be useful for testing SWRL mappings as well as running queries that should not be stored in the ontology.
In general, queries over mapped ontologies can be performed either directly on data materialised and stored in the core ontology, or by reformulating a core ontology query as a set of queries over the data stored in the source ontologies. Within rule-based mediation, SQWRL queries are executed when subsets of the knowledge stored within the core ontology need to be extracted.
2 Source Ontologies
In rule-based mediation, all ontologies which move data in and out of the core ontology are called source ontologies. Information flow between source and core ontologies is bi-directional and can be transitive, as translation between data sources via the core ontology is also possible. This section describes the source ontologies used to implement rule-based mediation for the model annotation use cases in Section 5.
A source ontology can be either a purpose-built syntactic ontology or a pre-existing ontology. Pre-existing source ontologies have no expressivity restrictions; community developed or other pre-existing ontologies which represent a field only tangentially related to the domain of interest can be used without modification. Mapping an existing ontology requires only a set of SWRL rules to connect it to the core ontology.
Information from one or more non-OWL data sources sharing the same format can be recast as a syntactic ontology, isolating syntactic variability so the core ontology is free to model the semantics of the relevant biology. Most syntactic ontologies are intended to be a useful representation of a format in OWL. Such a representation should be semantically lightweight and simple and quick to build. Strictly, it might be argued that syntactic ontologies are not true ontologies, being more precisely defined as a list of axioms. This is because syntactic ontologies are built as a simple conversion of a non-OWL format into OWL, with no additional semantics. There is little reason to enrich the syntactic ontologies when the majority of the inference, reasoning and querying will take place within the core ontology. Syntactic ontologies are a type of application ontology intended, not to be a reference for a particular biological domain of interest, but to facilitate a larger bioinformatics application.
Four data sources were used for the implementation of rule-based mediation presented in this chapter: BioGRID [26], exported in PSI-MIF [27]; the Pathway Commons [28] BioPAX Level 2 export; BioModels [29], in SBML format; and UniProtKB [30] in XML format. For each data format, a suitable source ontology was identified or syntactic ontology was created. BioModels was the data source for the SBML models to be annotated, while the other three resources were used as general data inputs to the integration system. Basic information on the data formats as well as the numbers of classes, relations and mapping rules in their source ontologies is available in Table 1.
2.1 Pathway Commons and BioPAX
Two data sources, UniProtKB and Pathway Commons, provide a data export in a format accessible to SWRL and OWL. For BioPAX, the creation of a specific syntactic ontology was not required. For the UniProtKB, XML rather than RDF was chosen for the reasons outlined in Section below.
Pathway Commons integrates nine pathway and interaction databases (Note: http://www.pathwaycommons.org/pc/dbSources.do). Information on network neighbours of any given entity can be accessed as a BioPAX Level 2 document, written in OWL. Pathway Commons provides both binary interaction data and pathway data. BioPAX can store information about which species are reactants, products or modifiers, but the data coming from Pathway Commons does not always provide this information. As this information is available in BioPAX, no additional syntactic ontology needs to be created; the populated BioPAX ontology returned from the nearest neighbour query is the source ontology.
2.2 Novel syntactic ontologies
BioModels and BioGRID do not export their data in a format amenable to the application of SWRL rules, and therefore syntactic ontologies were created based on the export format to which each data source was best suited. This required the use of MFO for BioModels entries and the creation of a syntactic ontology based on PSI-MIF. Additionally, the UniProtKB XML format needed to be converted to OWL as the RDF export was deemed inappropriate for the use cases.
The use of existing tools to implement rule-based mediation increases its usability for other researchers as well as decreases the development time. Protégé 3.4 and 4 were used to edit and view all ontologies used in this project, while the syntactic ontologies were generated using the XMLTab plugin [31] for Protégé 3.4. Once the syntactic ontologies are created, further editing may be performed as needed. Specifically, MFO was modified beyond the simple conversion created by XMLTab, as described in Chapter 4.
The XMLTab plugin takes only a few seconds to create syntactic ontologies. Additionally, if an XML file is provided instead of an XSD, then both classes and instances are generated: the classes represent structural elements and attributes present in the XML file, while the actual data is created as instances within the ontology. The resulting OWL file is an exact duplicate of the XML structure, which is acceptable for the rule-based mediation methodology as semantic heterogeneity is resolved when the data is mapped from the syntactic ontology to the core ontology.
However, XMLTab does not scale well, and cannot load multiple XML files into a single syntactic ontology. Additionally, it is not in active development. However, tools such as the SWRLAPI’s XMLMapper [32] or Krextor [33] may provide viable alternatives. Quality assurance and normalisation is not relevant with most syntactic ontologies, as they are direct syntax conversions and do not bear the greatest reasoning load in rule-based mediation. As such, the most important feature is that they are quick to generate and easy to produce, rather than being highly expressive or optimized for reasoning.
Model Format OWL
MFO is an ontology representing constraints from SBML, SBO and the SBML Manual [34] (see also Chapter 4). MFO is used as one of the source ontologies, and has a dual purpose for the presented use cases: firstly, it acts as the format representation for any data stored as SBML such as entries from the BioModels database, and secondly it is used to format query responses as SBML.
BioModels is an example of a single data source that may be loaded into a core ontology from more than one source ontology. BioModels entries are available in SBML format as well as in BioPAX and CellML. However, BioModels uses SBML as its native format, and conversion from SBML to other formats is not lossless. For example, quantitative data such as rate laws are not supported in BioPAX. Therefore when manipulating BioModels data, SBML—and therefore MFO—is the most suitable.
PSI-MIF
The PSI-MIF syntactic ontology was created from its native XML. BioGRID stores 24 different types of interactions. For the purposes of this work, its physical protein-protein interactions were used. BioGRID data is available in PSI-MI format (known as PSI-MIF) [27], and can easily be converted into a simple syntactic ontology. The PSI-MIF file exported by BioGRID was used to generate a simple syntactic ontology using XMLTab in Protégé 3.4. Figure 4 shows an overview of the classes created for the PSI-MIF syntactic ontology, while Figure 5 shows a selection of the axioms applied to the PSI-MIF interaction class.
As this work progressed, Pathway Commons began importing BioGRID data and can therefore can be used to retrieve BioGRID data in BioPAX format rather than requiring the PSI-MIF syntactic ontology. However, Pathway Commons does not store the genetic interactions available from BioGRID. The incorporation of BioGRID data into Pathway Commons does not reduce the effectiveness of the PSI-MIF syntactic ontology; the PSI-MIF syntactic ontology can be used to integrate any other PSI-MIF-based data sources, and it can also be used to integrate those portions of BioGRID which are not stored within Pathway Commons. The integration discussed in the use cases in Section 5 made use of the PSI-MIF syntactic ontology, while a Web application currently under development uses the BioPAX output from Pathway Commons.
UniProtKB
The UniProtKB is a comprehensive public protein sequence and function database, consisting both of manually-curated and automatically-annotated data. Adding UniProtKB as a data source provides access to a wealth of information about proteins, although the reaction data is more limited. As evidenced by the non-curated models stored within the BioModels database, new systems biology models are often created with only a small amount of biological information such as species names and skeleton reactions. In these cases, even the simple addition of cross-references to UniProtKB is useful. Unambiguous assignment of a UniProtKB primary accession to a new species in a systems biology model also provides the core ontology with high-quality knowledge of protein localization and identification.
While the original intent was to make use of the UniProtKB RDF format for rule-based mediation, reasoning over the RDF was slow and there was a large number of ontology and RDF files to import. Instead, the UniProtKB XML format was used for the initial work. At the beginning of this research, UniProtKB Release 14.8 XML files appropriate for the use cases were downloaded (Note: ftp://ftp.uniprot.org/pub/databases/uniprot/knowledgebase/). The UniProtKB syntactic ontology was created using the XMLTab plugin for Protégé 3.4. Figure 6 shows an excerpt of this ontology. The UniProtKB syntactic ontology was successfully used for the initial use cases detailed in Section 5.
A rule-based mediation Web application is being developed, where the set of relevant entries will be expanded from that required for the use cases to the entire UniProtKB database. As these larger sets of XML files can no longer easily be manually converted via the XMLTab plugin, the UniProtKB RDF syntax was revisited as a data source for the rule-based mediation Web application. The UniProtKB RDF format, together with the structure provided by the UniProtKB OWL ontology, is suitable for the application. The RDF syntax is accessible to SWRL rules without modification, and the slow reasoning times are irrelevant as reasoning over syntactic ontologies is not required with rule-based mediation. However, limitations remain in the UniProtKB RDF format.
Firstly, the core UniProtKB OWL file is not guaranteed to be decidable. This is because its expressivity level is OWL Full and not one of the less expressive, decidable profiles such as OWL DL. If an ontology is not decidable, there is no guarantee that a reasoner can successfully be run over the ontology. The expressivity level, or the OWL profile, of the core UniProtKB OWL file (Note: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/rdf/core.owl, file downloaded and test performed on 16 October 2011.) was tested using both the Manchester OWL Validator (Note: http://owl.cs.manchester.ac.uk/validator/) and the WonderWeb OWL Ontology Validator (Note: http://www.mygrid.org.uk/OWL/Validator). The former stated that, while the UniProtKB OWL file conformed to OWL 2, it did not conform any decidable profiles of OWL 2. The WonderWeb validator produced a similar result, stating that the file was OWL Full.
Secondly, the full model of the UniProtKB structure is stored in core file plus a further nine files, all ten of which must be imported to have a complete model (Note: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/rdf/README). The nine non-core files describe those parts of the UniProtKB structure which might be reused across multiple UniProtKB entries, and include topics such as citations, keywords and pathways. As each of these files contain all of the entities required by the entire UniProtKB dataset, many are quite large. This design requires the import of all ten files to completely view or process just one UniProtKB entry, creating a heavyweight method of modelling. For example, the RDF document containing all citations is 1.4 Gigabytes in size, making it difficult for many users to even load the document into memory for access by a reasoner.
3 Mapping between source and core ontologies
Once a source ontology has been created, it must be mapped to the core, biologically-relevant ontology. The core ontology (Section 4) is the mediator for rule-based mediation, and holds all of the biologically-meaningful statements that need to be made about the data to be integrated.
The mappings between the source ontologies and the core ontology are not as straightforward as the conversion from a data format to its syntactic ontology, and they are not created in the same way. The mapping in rule-based mediation is defined with the OWL-based SWRL rule language, which divides rules into a antecedent for describing the initial conditions of the rule and a consequent for describing the target of the rule (see Section 1.4). When mapping from the source ontologies to the core ontology, as a general rule the antecedent of the rule comes from a source ontology while the consequent corresponds to the mapped concept within the core ontology. The antecedent and consequent can be named classes or non-atomic classes such as those expressed by properties. In each of the rules presented in this section, the right arrow is a convenience rather than standard OWL notation, and the namespaces for each entity denote which ontology or library they belong to. For example, in the rule below, the antecedent is the protein class from the UniProtKB syntactic ontology, and the consequent is the Protein class from the telomere ontology (Note: The “tuo” namespace for the telomere ontology is a result of an earlier naming scheme, where the telomere ontology was called the telomere uncapping ontology.):
UP_00001 : upkb:protein(?protein)  tuo:Protein(?protein)
tuo:Protein(?protein)
Many of these rules could be merged together, having one rule perform much of the mapping currently executed by many. However, that would reduce the flexibility now present in the system. With each rule performing just one part of the mapping, each mapping step may be run in isolation. Further, having each rule accomplish just one task aids understanding and readability of the rules for humans. Readability is also aided with the addition of comments for each rule within the SWRL mapping file.
Once instances have been integrated into the core ontology, the knowledge contained within the core ontology can be reasoned over and queried. SQWRL is used to query the core ontology, and the query response can then be formatted according to a suitable source ontology. Queries are used to illustrate interesting, but only indirectly-relevant queries without cluttering the core ontology. More precise mapping rules use SWRL to allow storage of the results within the core ontology. The mapping between source and core ontologies can be bi-directional, thus allowing output of information as well as input. The quality of the data exported to a source ontology and from there to a base format is dependent upon both the quality of the mappings to the source ontology and its scope: if either the mappings are incomplete, or the underlying data format has no way of describing the new data, the export will be lacking.
3.1 Rules for the UniProtKB syntactic ontology
This section describes the SWRL rules of the UniProtKB syntactic ontology and what sort of information those rules map to the telomere ontology. The UniProtKB rule set is different from the other data sources’ rules. While UniProtKB contains a large amount of information, it is protein-centric rather than reaction-centric. More importantly for these use cases rich information about a protein, described in other databases with much less detail, is available to the telomere ontology via this data source. Specifically, information regarding protein localization within the cell as well as synonyms of gene and protein names are available. This rule set is simple, with many of the classes required for the use cases having direct equivalents in telomere ontology, and with many cardinalities of the properties exactly matching those in the telomere ontology itself. While the UniProtKB does contain some limited information on reactions via the comment and cross-reference sections, these are not currently modelled by the mapping rules for the UniProtKB syntactic ontology.
Protein identification
The first three rules in the UniProtKB syntactic ontology populate the Protein class with the protein instance itself as well as with a primary name and synonym:
UP_00001 :
upkb:protein(?protein) →
tuo:Protein(?protein)
UP_00002 :
upkb:recommendedNameSlot(?protein, ?name) ∧
upkb:fullName(?name, ?fullName) →
tuo:recommendedName(?protein, ?fullName)
UP_00003 :
upkb:entry(?entry) ∧
upkb:proteinSlot(?entry, ?protein) ∧
upkb:geneSlot(?entry, ?gene) ∧
upkb:nameSlot(?gene, ?geneName) ∧
upkb:Text(?geneName, ?geneNameText) →
tuo:synonym(?protein, ?geneNameText)
As it is a primary data source for protein information, UniProtKB is the only source ontology which populates the recommendedName property within the telomere ontology. All other data sources are mapped to the synonym data property. In UP_00003, the first part of the rule contains only the statement entry(?entry). From a computational point of view, this extra statement is superfluous, as the next part of the rule (proteinSlot(?entry, ?protein)) ensures the correct type of ?entry instance is used. However, the extra statement in this and other rules aids readability of those rules.
Protein localisation
If the location of a protein is the nucleus, then the instance is mapped directly to the Nucleus class. Otherwise, the instance is mapped to the more generic BiologicalLocalization class:
UP_00004 :
upkb:locationSlot(?subcellularLocation, ?location) ∧
upkb:Text(?location, ?locationText) ∧
swrlb:equal(?locationText, “Nucleus”) →
tuo:Nucleus(?location)
UP_00011 :
upkb:locationSlot(?subcellularLocation, ?location) ∧
upkb:Text(?location, ?locationText) ∧
swrlb:notEqual(?locationText, “Nucleus”) →
tuo:BiologicalLocalization(?location)
The two rules above use SWRL built-ins. The swrlb:equal and swrlb:notEqual built-ins test whether two arguments are equivalent. The test for equality is satisfied if and only if the first argument and the second argument are identical, while the swrlb:notEqual is the simple negation of swrlb:equal [17].
As well as creating instances for each biological location, linking those locations to a specific protein also occurs. In UP_00006, the subcellular location of the protein is mapped directly onto the isLocated property within the telomere ontology.
UP_00006 :
upkb:entry(?entry) ∧
upkb:proteinSlot(?entry, ?protein) ∧
upkb:commentSlot(?entry, ?comment) ∧
upkb:subcellularLocationSlot(?comment, ?subcellularLocation) ∧
upkb:locationSlot(?subcellularLocation, ?location) →
tuo:isLocated(?protein, ?location)
Taxonomic identification and other database cross-references
The next three rules describe how the cross references of a protein are mapped from UniProtKB to the telomere ontology. UP_00005 creates an instance of TaxonomicSpecies with a specific taxonomic identifier. Conversely, UP_00007 is used to direct all other UniProtKB references to the more generic DatabaseReference from the telomere ontology. Both swrlb:equal and swrlb:notEqual have been used in these rules to determine whether or not a particular reference is a taxonomic identifier. At this stage, the information is not yet connected to a protein. UP_00008 provides this functionality, linking a particular protein with its taxonomic reference.
UP_00005 :
upkb:dbReference(?reference) ∧
upkb:_type(?reference, ?referenceType) ∧
swrlb:equal(?referenceType, “NCBI Taxonomy”) ∧
upkb:_id(?reference, ?id) →
tuo:TaxonomicSpecies(?reference) ∧
tuo:ncbiTaxId(?reference, ?id)
UP_00007 :
upkb:dbReference(?reference) ∧
upkb:_type(?reference, ?referenceType) ∧
upkb:_id(?reference, ?id) swrlb:notEqual(?referenceType, “NCBI Taxonomy”) →
tuo:DatabaseReference(?reference) ∧
tuo:databaseName(?reference, ?referenceType) ∧
tuo:accession(?reference, ?id)
UP_00008 :
upkb:entry(?entry) ∧
upkb:proteinSlot(?entry, ?protein) ∧
upkb:organismSlot(?entry, ?organism) ∧
upkb:dbReferenceSlot(?organism, ?reference) →
tuo:hasTaxon(?protein, ?reference)
UP_00009 has a structure virtually identical to UP_00008, but rather than mapping the organism references for a particular protein instance, it maps all associated database references to their equivalents in the telomere ontology.
UP_00009 :
upkb:entry(?entry) ∧
upkb:proteinSlot(?entry, ?protein) ∧
upkb:dbReferenceSlot(?entry, ?reference) →
tuo:hasDatabaseReference(?protein, ?reference)
UP_00010 uses a SWRL built-in called swrlx:makeOWLThing to create a new DatabaseReference for each UniProtKB entry. Rather than being part of the W3C Submission, built-ins with the namespace swrlx are experimental convenience methods. swrlx:makeOWLThing takes an empty variable as its first argument, and will create and bind a new instance to that variable for every instance of the second argument matched within the rule (Note: http://protege.cim3.net/cgi-bin/wiki.pl?SWRLExtensionsBuiltIns). The SWRL built-in does not fill the value of the first argument with the actual instance or instances present in later arguments; it simply uses the second and later arguments as a statement of how many new instances to create. The associated UniProtKB accession number is used as the value for the new instance’s accession data property, while the database reference name is given as “UniProtKB”.
UP_00010 :
upkb:entry(?entry) ∧
upkb:proteinSlot(?entry, ?protein) ∧
upkb:accession(?entry, ?accessionValue) ∧
swrlx:makeOWLThing(?reference, ?entry) ∧
swrlb:equal(?referenceValue, “UniProtKB”) →
tuo:hasDatabaseReference(?protein, ?reference) ∧
tuo:DatabaseReference(?reference) ∧
tuo:accession(?reference, ?accessionValue) ∧
tuo:databaseName(?reference, ?referenceValue)
3.2 Rules for the PSI-MIF syntactic ontology
This section describes the SWRL rules of the PSI-MIF syntactic ontology, and what sort of information those rules map to the telomere ontology. This ontology is centred around binary interactions, with some extra information provided regarding taxonomy and database cross-references. Unlike BioPAX, BioGRID provides only binary interactions, and as such the information modelled in the BioGRID syntactic ontology is only of this interaction type.
One-to-one mappings
As with BioPAX, there is a high degree of convergence between the telomere ontology and BioGRID. BioPAX, BioGRID and the telomere ontology all model interactions in a similar way. This is exemplified by the five classes within the BioGRID syntactic ontology which are mapped directly to classes within the telomere ontology. PSIMIF_00001 is shown here, and PSIMIF_00003-6 are described in later subsections.
PSIMIF_00001 :
psimif:interactor(?interactor) →
tuo:PhysicalEntity(?interactor)
The convergence is further visible in rules PSIMIF_00002, PSIMIF_00010 and PSIMIF_00012-14 (see subsections below), where the classes and the properties described have direct one-to-one mappings with classes and properties in the telomere ontology. Although the one-to-one mapping rules could be accomplished with standard OWL axioms, it is useful to keep all cross-ontology mapping logically separate from the telomere ontology itself. Not only does this allow changes to the rules without changing the telomere ontology itself, but it also collects all mappings in one location for improved readability.
Identification
Two rules in the BioGRID syntactic ontology populate the Protein class with instance data as well as with synonyms. Unlike UniProtKB, which only stores protein entities, BioGRID can have a number of different types of interactor. Therefore, in order to only retrieve those interactors which are proteins, PSIMIF_00015 is restricted to those interactor instances which are of the “protein” type. Note that no recommendedName is populated in the telomere ontology via the BioGRID syntactic ontology; only the UniProtKB syntactic ontology provides information for that property. All other source ontologies provide information for the synonym data property only.
PSIMIF_00015 :
psimif:fullName(?name, ?nameText) ∧
swrlb:equal(?nameText, “protein”) ∧
psimif:interactor(?interactor) ∧
psimif:interactorTypeSlot(?interactor, ?interactorType) ∧
psimif:namesSlot(?interactorType, ?name) →
tuo:Protein(?interactor)
PSIMIF_00011 :
psimif:interactor(?interactor) ∧
psimif:namesSlot(?interactor, ?names) ∧
psimif:shortLabel(?names, ?nameText) →
tuo:synonym(?interactor, ?nameText)
Taxonomic identification and other database cross-references
The two rules below assign the taxonomy instance from BioGRID to the telomere ontology TaxonomicSpecies, and then assign the mapped taxonomic instance to the appropriate interactor. In PSIMIF_00002, as in other rules, object and data properties for all PSI-MIF interator instances are mapped rather than just those for protein instances, as in PSIMIF_00015. While this will generate more properties within the telomere ontology then are required, no extraneous PhysicalEntity instances are created. General-purpose rules such as these are simpler, and do not contribute to a large increase in computational complexity.
PSIMIF_00002 :
psimif:organism(?organism) ∧
psimif:_ncbiTaxId(?organism, ?taxId) →
tuo:TaxonomicSpecies(?organism) ∧
tuo:ncbiTaxId(?organism, ?taxId)
PSIMIF_00010 :
psimif:interactor(?interactor) ∧
psimif:organismSlot(?interactor, ?organism) →
tuo:hasTaxon(?interactor, ?organism)
All non-taxonomic cross-references are identified in PSI-MIF with the primaryRef or secondaryRef classes. These, and the names of the databases themselves, are mapped to the telomere ontology’s DatabaseReference class in rules PSIMIF_00005-6 and PSIMIF_00012. Finally, in PSIMIF_00013-14 the identifiers of each of the individual cross-references are mapped to the accession property in the telomere ontology.
PSIMIF_00005 :
psimif:primaryRef(?reference) →
tuo:DatabaseReference(?reference)
PSIMIF_00006 :
psimif:secondaryRef(?reference) →
tuo:DatabaseReference(?reference)
PSIMIF_00012 :
psimif:_db(?reference, ?referenceName) →
tuo:databaseName(?reference, ?referenceName)
PSIMIF_00013 :
psimif:primaryRef(?primaryReference) ∧
psimif:_id(?primaryReference, ?id) →
tuo:accession(?primaryReference, ?id)
PSIMIF_00014 :
psimif:secondaryRef(?secondaryReference) ∧
psimif:_id(?secondaryReference, ?id) →
tuo:accession(?secondaryReference, ?id)
After the cross-references are mapped, they need to be tied to the individual PhysicalEntity instances which reference them. The link between a cross-reference and a PSI-MIF entity occurs with the xrefSlot property and either the primaryRefSlot or the secondaryRefSlot. These properties are simplified and mapped across to the hasDatabaseReference property in the telomere ontology.
PSIMIF_00016 :
psimif:xrefSlot(?entity, ?crossRef) ∧
psimif:primaryRefSlot(?crossRef, ?primaryRef) →
tuo:hasDatabaseReference(?entity, ?primaryRef)
PSIMIF_00017 :
psimif:xrefSlot(?entity, ?crossRef) ∧
psimif:secondaryRefSlot(?crossRef, ?secondaryRef) →
tuo:hasDatabaseReference(?entity, ?secondaryRef)
Binary interactions
Although less sophisticated than BioPAX, BioGRID defines interactions in a similar way. The participant in a binary interaction and the physical entity that is referenced by that participant are two separate entities. However, unlike BioPAX, BioGRID reactions are only expressed as binary interactions of varying types, with no concept of complex reactions containing multiple inputs or outputs. Additionally, there is no way to determine which entity is a product or modifier of the interaction. Therefore, PSIMIF_00003 maps all participants in the binary interactions to the telomere ontology’s Reactant class. In PSIMIF_00004 all interactions, of whatever type, are mapped to the telomere ontology’s PhysicalProcess class. In PSIMIF_00007 the instances of the “ReconstitutedComplex” interactions are then further mapped to the ProteinComplexFormation class in the telomere ontology.
PSIMIF_00003 :
psimif:participant(?participant) →
tuo:Reactant(?participant)
PSIMIF_00004 :
psimif:interaction(?interaction) →
tuo:PhysicalProcess(?interaction)
PSIMIF_00007 :
psimif:interaction(?interaction) ∧
psimif:interactionTypeSlot(?interaction, ?interactionType) ∧
psimif:namesSlot(?interactionType, ?interactionName) ∧
psimif:shortLabel(?interactionName, ?nameText) ∧
swrlb:equal(?nameText, “ReconstitutedComplex”) →
tuo:ProteinComplexFormation(?interaction)
The link between the interactor and its participant in BioGRID is modeled with the interactorRef property. In the telomere ontology, this connection is made with the plays property:
PSIMIF_00008 :
psimif:interactor(?interactor) ∧
psimif:_id(?interactor, ?id) ∧
psimif:participant(?participant) ∧
psimif:interactorRef(?participant, ?id) →
tuo:plays(?interactor, ?participant)
Finally, PSIMIF_00009 is used to link an interaction to its participants:
PSIMIF_00009 :
psimif:participantListSlot(?interaction, ?list) ∧
psimif:participantSlot(?list, ?participant) →
tuo:hasReactant(?interaction, ?participant)
3.3 Rules for BioPAX Level 2
One-to-one mapping and interactions
As described in Section 2.1, Pathway Commons provides information in BioPAX Level 2 format. The simplicity of the mapping between BioPAX Level 2 and the telomere ontology is due to the similar modelling of physical processes within them. As described in Section 3.2 with respect to other simple mappings, rules such as BP_00001-5 could be written in pure OWL but are written in SWRL to keep rules separate from ontology modelling. These rules identify the direct equivalents in the telomere ontology for each relevant class and property within BioPAX. Specifically, BP_00001-5 provide mappings for BioPAX proteins, reaction participants and interactions.
BP_00001 :
bp:protein(?protein) →
tuo:Protein(?protein)
BP_00002 :
bp:physicalEntityParticipant(?participant) →
tuo:Participant(?participant)
BP_00004 :
bp:physicalInteraction(?interaction) →
tuo:PhysicalProcess(?interaction)
BP_00003 :
bp:PHYSICAL-ENTITY(?participant, ?physicalEntity) →
tuo:playedBy(?participant, ?physicalEntity)
BP_00005 :
bp:PARTICIPANTS(?interaction, ?participant) →
tuo:hasReactant(?interaction, ?participant)
Protein identification
Rules BP_00008-9 map the name and synonym data properties present within a BioPAX protein to the telomere ontology synonym data property. As described in Section 3.1, only UniProtKB entries populate the recommendedName data property; all names and synonyms from other source ontologies map directly to the synonyms within the telomere ontology.
BP_00008 :
bp:protein(?protein) ∧
bp:NAME(?protein, ?value) →
tuo:synonym(?protein, ?value)
BP_00009 :
bp:protein(?protein) ∧
bp:SYNONYMS(?protein, ?value) →
tuo:synonym(?protein, ?value)
Taxonomic identification and other database cross-references
BP_00006 fills the TaxonomicSpecies class as well as the ncbiTaxId data property within the telomere ontology. In order to fill this value, only those unificationXref classes with a database whose name is “NCBI” are used. The identifier for this unificationXref is then used to fill the ncbiTaxId property. BP_00007 then links a Protein in the telomere ontology to its taxonomic species.
BP_00006 :
bp:unificationXref(?xref) ∧
bp:DB(?x, ?value) ∧
bp:ID(?x, ?id) ∧
swrlb:equal(?value, “NCBI”) →
tuo:TaxonomicSpecies(?xref) ∧
tuo:ncbiTaxId(?xref, ?id)
BP_00007 :
bp:protein(?protein) ∧
bp:ORGANISM(?protein, ?organism) ∧
bp:TAXON-XREF(?organism, ?ref) →
tuo:hasTaxon(?protein, ?ref)
BP_00010 fills the DatabaseReference class in the telomere ontology as well as the properties connecting the DatabaseReference to a database name and accession. Taxonomic identifiers are specifically not included in this rule, as they have already been parsed by BP_00006-7. BP_00011 then links the protein to the mapped DatabaseReference class via the hasDatabaseReference property. unificationXref instances are the only ones mapped from BioPAX, as they show equivalence between the BioPAX entity and the database reference specified. There are other reference types within BioPAX, however such cross references link to data that is related, but not identical, to the entity in question.
BP_00010 :
bp:unificationXref(?xref) ∧
bp:DB(?xref, ?dbvalue) ∧
bp:ID(?xref, ?idvalue) ∧
swrlb:notEqual(?dbvalue, “NCBI”) →
tuo:DatabaseReference(?xref) ∧
tuo:databaseName(?xref, ?dbvalue) ∧
tuo:accession(?xref, ?idvalue)
BP_00011 :
bp:protein(?protein) ∧
bp:XREF(?protein, ?xref) ∧
bp:unificationXref(?xref) →
tuo:hasDatabaseReference(?protein, ?xref)
3.4 Rules for MFO
There is a clear delineation in most systems biology formalisms between a protein as an abstract concept and a protein as a specific interactor in a reaction. Within SBML, this is defined using the speciesType or species elements for the concept of a protein, and speciesReference for the participant in the reaction. Within BioPAX Level 2, the biological entity / participant pairing is modelled with physicalEntity and physicalEntityParticipant. Within BioGRID, interactor and participant elements are used.
A similar dichotomy exists within the telomere ontology, and when converting the information retrieved in Use Case 1 back to SBML, these considerations need to be taken into account. One solution is to link the telomere ontology’s Protein class (and its parent class PhysicalEntity) to the SBML speciesType element. However, most SBML models do not use speciesType, and future versions of SBML will have a different way of modelling such concepts [35]. Therefore, to align with future versions of SBML as well as to make the mapping simple, the instances of the telomere ontology PhysicalEntity class are instead linked to the appropriate attributes of an SBML species element.
Biological annotation
MIRIAM annotation is added to MFO by mapping a variety of database cross-references from the telomere ontology. The rules used to create these annotations introduce two additional SWRL built-ins, swrlb:stringEqualIgnoreCase and swrlb:stringConcat. The swrlb:stringEqualIgnoreCase built-in is satisfied if and only if the two arguments match in a case-insensitive manner, while the first argument in swrlb:stringConcat is filled with a concatenation of all further arguments [17].
In the first two rules for mapping information from the telomere ontology to MFO (TUO_MFO_00001 and TUO_MFO_00001_synonym), all Rad9 instances are enhanced with two biological annotations: firstly, a MIRIAM URI for the UniProtKB entry associated with the Rad9 protein and secondly, the addition of a link to the SBO “macromolecule” term (SBO_0000245) (Note: For brevity, the second rule (TUO_MFO_00001_synonym) is not shown. It is identical to the first rule other than the swrlb:stringEqualIgnoreCase compares against “uniprotkb” rather than “uniprot”. Different source datasets use different names for this database.). This association is double-checked by ensuring that the “rad9” species name is already present. This is a rule which returns information—required for the use cases—on the Rad9 protein only. While this is a highly specific rule, it can easily be generalised to run over Protein, the parent class of Rad9. In such a general case, shown later for TUO_MFO_00006 and TUO_MFO_00006_synonym, all of the references to specific MFO species instances have been removed.
TUO_MFO_00001 :
tuo:Rad9(?protein) ∧
tuo:hasDatabaseReference(?protein, ?crossref) ∧
tuo:databaseName(?crossref, ?dbname) ∧
tuo:accession(?crossref, ?accession) ∧
swrlb:stringEqualIgnoreCase(?dbname, “uniprot”) ∧
swrlb:stringConcat(?miriamIs, “urn:miriam:uniprot:”, ?accession) ∧
sbo:SBO_0000245(?term) ∧
mfo:Species(?mfoSpecies) ∧
mfo:name(?mfoSpecies, ?nameText) ∧
swrlb:stringEqualIgnoreCase(?nameText, “rad9”) →
mfo:MiriamAnnotation(?crossref) ∧
mfo:miriamAnnotation(?mfoSpecies, ?crossref) ∧
mfo:bqbIs(?crossref, ?miriamIs) ∧
mfo:sboTerm(?mfoSpecies, ?term)
Additional MIRIAM annotations are added with TUO_MFO_00002, which adds any SGD cross-references to MFO for Rad9 instances. As with TUO_MFO_00001 and TUO_MFO_00001_synonym, this rule could easily be made more generic to allow all MFO species to be annotated with their relevant cross-references. As MIRIAM URIs require a prefix identifying the referenced database (e.g. “urn:miriam:uniprot:”), each database requires its own rule to correctly build the URI. Rules TUO_MFO_00003-4 (not shown) differ from TUO_MFO_00001-2 only in the database used to create the new MIRIAM annotation; TUO_MFO_00003 for Intact and TUO_MFO_00004 for Pathway Commons.
TUO_MFO_00002 :
tuo:Rad9(?protein) ∧
tuo:hasDatabaseReference(?protein, ?crossref) ∧
tuo:databaseName(?crossref, ?dbname) ∧
tuo:accession(?crossref, ?accession) ∧
swrlb:stringEqualIgnoreCase(?dbname, “sgd”) ∧
swrlb:stringConcat(?miriamIs, “urn:miriam:sgd:”, ?accession) ∧
mfo:Species(?species) ∧
mfo:name(?species, ?nameText) ∧
swrlb:stringEqualIgnoreCase(?nameText, “rad9”) →
mfo:MiriamAnnotation(?crossref) ∧
mfo:miriamAnnotation(?species, ?crossref) ∧
mfo:bqbIs(?crossref, ?miriamIs)
In TUO_MFO_00006 and TUO_MFO_00006_synonym, for every Protein in the telomere ontology, a UniProtKB MIRIAM annotation is created. (Note: TUO_MFO_00006_synonym is not shown in this section as it is identical to TUO_MFO_00006 except for the string comparison swrlb:stringEqualIgnoreCase, which instead uses the literal “uniprotkb”). In addition to the UniProtKB MIRIAM annotations within MFO for each telomere ontology Protein, TUO_MFO_00007-9 add MIRIAM annotations for SGD, Intact and Pathway Commons. Due to their similarity with TUO_MFO_00006, for brevity they are not shown.
TUO_MFO_00006 :
tuo:Protein(?mfoSpecies) ∧
mfo:Species(?mfoSpecies) ∧
tuo:hasDatabaseReference(?mfoSpecies, ?crossref) ∧
tuo:databaseName(?crossref, ?dbname) ∧
tuo:accession(?crossref, ?accession) ∧
swrlb:stringEqualIgnoreCase(?dbname, “uniprot”) ∧
swrlb:stringConcat(?miriamIs, “urn:miriam:uniprot:”, ?accession) ∧
sbo:SBO_0000245(?term) →
mfo:MiriamAnnotation(?crossref) ∧
mfo:miriamAnnotation(?mfoSpecies, ?crossref) ∧
mfo:bqbIs(?crossref, ?miriamIs) ∧
mfo:sboTerm(?mfoSpecies, ?term)
TUO_MFO_00011-12 provide name properties for the Species instances within MFO. As SWRL rules will not overwrite existing name values, this rule will only modify the mapped name property if it is empty. This ensures that existing values are not overwritten, and further ensures—if the rules are run in sequence—that names from the recommendedName property are not overridden by those from the synonym property. If pre-existing names from the MFO model should be re-written, they can simply be deleted prior to or during conversion from SBML to MFO.
TUO_MFO_00011 :
tuo:Protein(?spec) ∧
mfo:Species(?spec) ∧
tuo:recommendedName(?spec, ?recName) →
mfo:name(?spec, ?recName)
TUO_MFO_00012 :
tuo:Protein(?spec) ∧
mfo:Species(?spec) ∧
tuo:synonym(?spec, ?synonym) →
mfo:name(?spec, ?synonym)
Rad9-Rad17 Interaction (TUO_MFO_00005)
Specific interactions relating to Rad9 were retrieved from the telomere ontology and mapped to MFO. Rad9–Rad17 is described in this section, and Rad9–Mec1 is described in the next. TUO_MFO_00005 maps the Rad9–Rad17 interaction, and is tailored to populating a single SBML model. Otherwise, if multiple models are stored within MFO, incorrect listOfReactions and listOfSpecies could be matched. Where necessary, this limitation can easily be resolved by extending the rule to specify the ListOf__ instances associated with a particular systems biology model.
As TUO_MFO_00005 is a long and complex rule, it is presented in sections rather than all at once. Further, in contrast to the native ordering in the OWL files themselves, portions of the rule have been reordered slightly for increased readability. Firstly the reaction to be mapped, the reactants involved, and the proteins associated with those reactants within the telomere ontology are identified:
TUO_MFO_00005 :
tuo:ProteinComplexFormation(?reaction) ∧
tuo:Rad9Rad17Interaction(?reaction) ∧
tuo:hasReactant(?reaction, ?reactant) ∧
tuo:playedBy(?reactant, ?protein) ∧
Next, the already-extant ListOfReactions and ListOfSpecies instances from MFO are stored in SWRL variables for later use:
mfo:ListOfReactions(?listOfReaction) ∧
mfo:ListOfSpecies(?listOfSpecies) ∧
SWRL built-ins are then called to create seven new variables. Firstly, ?nameVariable is filled with the value “Rad9Rad17Complex” using swrlb:stringConcat; this is a string literal only, and is not an instance itself. ?nameVariable is used later in the rule to create a value for the name data property within MFO. Six calls to swrlx:makeOWLThing are then run. These calls create the appropriate number of MFO instances for storing the information related to the reactions and proteins matched within this rule. For instance, if there are  instances of the Protein class matched within TUO_MFO_00005, then new instances are created and bound to the variable ?speciesId. As the Rad9–Rad17 interaction is a protein complex formation, one product—the complex itself—is created for each reaction. The newly-created instance variable ?product describes the new product of the reaction. The other new variables create additional parts of an SBML model which are not directly modelled within the telomere ontology.
instances of the Protein class matched within TUO_MFO_00005, then new instances are created and bound to the variable ?speciesId. As the Rad9–Rad17 interaction is a protein complex formation, one product—the complex itself—is created for each reaction. The newly-created instance variable ?product describes the new product of the reaction. The other new variables create additional parts of an SBML model which are not directly modelled within the telomere ontology.
swrlb:stringConcat(?nameVariable, “Rad9Rad17Complex”) ∧
swrlx:makeOWLThing(?reactantList, ?reaction) ∧
swrlx:makeOWLThing(?reactionSId, ?reaction) ∧
swrlx:makeOWLThing(?speciesSId, ?protein) ∧
swrlx:makeOWLThing(?product, ?reaction) ∧
swrlx:makeOWLThing(?prodSpecSid, ?reaction) ∧
swrlx:makeOWLThing(?prodRef, ?reaction) ∧
swrlx:makeOWLThing(?productList, ?reaction)
These SWRL built-ins mark the end of the antecedent for TUO_MFO_00005, which has created or identified all variables needed to map the reaction data to MFO. The consequent begins with assignment of all of the identifiers to SId instances in MFO.
→
mfo:SId(?reactionSId) ∧
mfo:SId(?speciesSId) ∧
mfo:SId(?prodSpecSid) ∧
Then, each of the links to those SId instances is built using the id object property. Additionally, the name property is filled for the ?product instance:
mfo:id(?reaction, ?reactionSId) ∧
mfo:id(?protein, ?speciesSId) ∧
mfo:id(?product, ?prodSpecSid) ∧
mfo:name(?product, ?nameVariable) ∧
Next, each of the instances named in the first argument of the id property are mapped to their appropriate MFO classes. Additionally, the Reaction instance gets a mapped list of reactants and products and both the Species and Reaction instances are linked to their appropriate ListOf__ instances.
mfo:Reaction(?reaction) ∧
mfo:Species(?protein) ∧
mfo:Species(?product) ∧
mfo:listOfReactants(?reaction, ?reactantList) ∧
mfo:listOfProducts(?reaction, ?productList) ∧
mfo:reaction(?listOfReaction, ?reaction) ∧
mfo:species(?listOfSpecies, ?protein) ∧
mfo:species(?listOfSpecies, ?product) ∧
The ListOfSpeciesReferences class in MFO is then populated with the appropriate new instances from the swrlx:makeOWLThing built-in, and each individual SpeciesReference is mapped and added to its list:
mfo:ListOfSpeciesReferences(?reactantList) ∧
mfo:ListOfSpeciesReferences(?productList) ∧
mfo:ProductSpeciesReference(?prodRef) ∧
mfo:ReactantSpeciesReference(?reactant) ∧
mfo:speciesReference(?reactantList, ?reactant) ∧
mfo:speciesReference(?productList, ?prodRef) ∧
Rad9-Mec1 Interaction (TUO_MFO_00010)
There are two interactions between Rad9 and Mec1 present within the telomere ontology. In this rule, one of these interactions has been arbitrarily chosen to send to MFO. This is a choice the modeller can make in future, after looking at the information contained in each interaction choice. Unlike TUO_MFO_00005, there is no extra information from the data sources which allows the interaction to be defined as being a protein complex formation. As such, a likely product for the reaction cannot be created. The lack of knowledge about the interaction means that TUO_MFO_00010 is a much simpler rule than TUO_MFO_00005. Except for those sections of the rule dealing with products and lists of products, TUO_MFO_00010 is the same as TUO_MFO_00005. As a result the reaction has reactants only, and no products.
TUO_MFO_00010 :
tuo:Rad9Mec1Interaction(’psimif:interaction_143’) ∧
tuo:hasReactant(’psimif:interaction_143’, ?react) ∧
tuo:playedBy(?react, ?protein) ∧
mfo:ListOfReactions(?listOfReaction) ∧
swrlx:makeOWLThing(?reactantList, ’psimif:interaction_143’) ∧
swrlx:makeOWLThing(?reactSId, ’psimif:interaction_143’) ∧
swrlx:makeOWLThing(?specSId, ?protein) →
mfo:ListOfSpeciesReferences(?reactantList) ∧
mfo:SId(?reactSId) ∧
mfo:SId(?specSId) ∧
mfo:ReactantSpeciesReference(?react) ∧
mfo:speciesReference(?reactantList, ?react) ∧
mfo:Reaction(’psimif:interaction_143’) ∧
mfo:id(’psimif:interaction_143’, ?reactSId) ∧
mfo:reaction(?listOfReaction, ’psimif:interaction_143’) ∧
mfo:Species(?protein) ∧
mfo:id(?protein, ?specSId) ∧
mfo:listOfReactants(’psimif:interaction_143’, ?reactantList)
4 The core ontology
Often, mediator-based approaches build a core ontology as a union of source ontologies rather than as a semantically-rich description of the research domain in its own right [36, 16, 37]. While such ontologies thoroughly describe the associated data formats, a description of the structure of data is not a description of the biology of the data. A union of source ontologies would not produce a description of the biological domain of interest, but simply a description of the combined formats imported into the system. Further, if a core ontology is defined as merely an ontology which models a set of data sources, the core ontology becomes brittle with respect to the addition of new data sources and new formats.
In contrast, for rule-based mediation a core ontology should be a biologically-relevant, tightly-scoped, logically-rigorous description of the semantics of the research domain. Although the definition of a core ontology shares much with that of a reference ontology, a core ontology is not required to stand as a reference model for a biological domain of interest, although that may be one of its additional purposes. As originally written about BioPAX, a useful representation of the research domain should be detailed enough to express richness where required, but general enough to maintain the overall framework when few details are available [38]. The core ontology may be created for rule-based mediation or drawn from existing ontologies. However, pre-existing biomedical ontologies should be chosen with care, as many do not have sufficiently formalised semantics and as such may not be useful for automated reasoning tasks in a data integration context [7]. A core ontology is not intended to capture all of biology; instead, it is scoped tightly to its purpose, which is modelling the research domain of interest.
By creating an core ontology which is more than the entailment of a set of source ontologies, and which stands on its own as a semantically-rich model of a research domain, the core ontology becomes much more flexible with respect to changes in data sources. Because the core ontology is abstracted away from data formats, importing new data sources is made easier. Furthermore, the process of adding to the core ontology is simplified: each new mapping, class, or data import is incremental, without the need for large-scale changes. The richness of the core ontology depends on the type of biological questions that it has been created to answer and the amount of resources available; a detailed ontology may have higher coverage of the research domain, but may take longer to develop and reason over.
In contrast to global-as-view or local-as-view approaches, where either the target or source ontologies are entirely described using views of the other, the methods used in rule-based mediation provide a way to decouple syntactic and semantic heterogeneity and allow the core and source ontologies to remain distinct. Additionally, rule-based mediation maps and loads data directly into the core ontology, allowing simplified querying via the use of standard tools, mapping and querying languages. Being a semantically-rich model of a research domain, a core ontology could also be used as a standalone ontology for marking up data or as a reference model of its biological domain.
It is possible for a core ontology to incorporate other domain ontologies or upper-level ontologies. Hoehndorf and colleagues included a custom-built upper-level ontology to aid the integration of the in vivo and in silico aspects of SBML models [14]. However, as rule-based mediation uses the source and core ontologies to deliberately keep the biology and data structure separate, such an upper-level ontology is not required. Upper-level ontologies are often created to aid in the merging of ontologies with distantly-connected topics. As such, they are not as important for core ontologies, which are tied to one specific subject.
4.1 The Telomere Ontology
For the use cases presented in this chapter the core ontology, referred to as the telomere ontology, models a portion of the telomere binding process in S.cerevisiae. Specifically, it models selected parts of the biology relevant to the Proctor and colleagues model [39] of telomere uncapping. Figure 7 shows an overview of this ontology.
The majority of the reasoning and querying in rule-based mediation is run over the integrated data in the core ontology, allowing appropriate annotations to be queried for and selected. The results of those queries can then be exported, via a source ontology, to a non-OWL format for that data source. The use cases described in Section 5 illustrate the retrieval of knowledge from the telomere ontology and its application to systems biology model annotation via the MFO syntactic ontology.
telomere ontology would be only be of the type DirectedReaction, this constraint does not need to be explicitly stated. If required, inferences about the data source itself could be made by examination of its instances in telomere ontology.
4.2 Rules and stored queries within the telomere ontology
There are a number of rules where both the antecedent and the consequent contain entities only from the telomere ontology. These are generally convenience methods to make statements about the data coming into the ontology which otherwise would be difficult to do in plain OWL. For instance, TUO_00001-2 further specialise instances of Protein to be instances of Rad9 if they have “rad9” somewhere in the recommended name or the synonym list. These rules are used within Use Case 1 to store the results of a query against the integrated dataset which places all RAD9 proteins within the Rad9 class (see Section 5.1). While these rules work for relatively small datasets, the false positive rate might increase as the datasets grow. Further work on the telomere ontology could replace these rules with more precise axioms defining the classes they are modifying. Optionally, automated instance-to-class mapping (see Section 6.4) would eliminate the need for specific rules such as these, as would modifying the way proteins are handled such that each protein (e.g. Rad9) is not given its own class.
TUO_00001 :
tuo:Protein(?someEntity) ∧
tuo:synonym(?someEntity, ?synonym) ∧
swrlb:containsIgnoreCase(?synonym,“rad9”) →
tuo:Rad9(?someEntity)
TUO_00002 :
tuo:Protein(?someEntity) ∧
tuo:recommendedName(?someEntity, ?name) ∧
swrlb:containsIgnoreCase(?name, “rad9”) →
tuo:Rad9(?someEntity)
TUO_00004-11 are similar rules for four other subclasses of Protein: Rad53, Chk1, Mec1 and Rad17. These include matching specific name such as “rad53” in TUO_00004, as well as other known synonym matches such as “YPL153C” in TUO_00004_Systematic. Only two of these rules are shown, with the others not included for brevity.
TUO_00004 :
tuo:Protein(?someEntity) ∧
tuo:synonym(?someEntity, ?synonym) ∧
swrlb:containsIgnoreCase(?synonym, “rad53”) →
tuo:Rad53(?someEntity)
TUO_00004_Systematic :
tuo:Protein(?someEntity) ∧
tuo:synonym(?someEntity, ?synonym) ∧
swrlb:containsIgnoreCase(?synonym, “YPL153C”) →
tuo:Rad53(?someEntity)
Other rules of this type were created specifically to perform the same task as the axioms present in the defined class referenced in the rules (see Use Case 2 in Section 5.2 for more information), thus allowing users of the ontology to see the results of the inference without needing to realise the ontology with a reasoner (see Section 1.5 for a description of realisation). An example of this is TUO_00012, which populates the Rad9Rad17Interaction and associated properties without needing to infer the placement of instances via a reasoner. Rules TUO_00013-15 (not shown) are virtually identical to TUO_00012, instead mapping the information required for Rad9Rad53Interaction, Rad9Mec1Interaction and Rad9Chk1Interaction.
TUO_00012 tuo:Rad9(?rad9instance) ∧
tuo:plays(?rad9instance, ?rad9participant) ∧
tuo:Rad17(?rad17instance) ∧
tuo:plays(?rad17instance, ?rad17participant) ∧
tuo:hasParticipant(?process, ?rad9participant) ∧
tuo:hasParticipant(?process, ?rad17participant) →
tuo:Rad9Rad17Interaction(?process)
Finally, there are two stored SQWRL queries in the telomere ontology. TUO_SQWRL_00001 selects those entities with a taxonomic identifier of 4932 and which are instances of the Rad9 class, which itself is filled via TUO_00001 and TUO_00002.
TUO_SQWRL_00001 :
tuo:Rad9(?someEntity) ∧
tuo:hasTaxon(?someEntity, ?x) ∧
tuo:ncbiTaxId(?x, ?y) ∧
swrlb:equal(?y, 4932) →
sqwrl:selectDistinct(?someEntity)
TUO_SQWRL_00004 retrieves only those reactions that involve Rad9, and is used within Use Case 2 to retrieve specific interactions with Rad9 instances (see Section 5.2).
TUO_SQWRL_00004 :
tuo:Rad9(?rad9instance) ∧
tuo:plays(?rad9instance, ?participant) ∧
tuo:hasParticipant(?process, ?participant) →
sqwrl:select(?rad9instance, ?process)
5 Two use cases demonstrate rule-based mediation
Rule-based mediation has been implemented for the two use cases presented in this section. A more generic Web implementation of rule-based mediation is currently under development. The use cases show how rule-based mediation extends a quantitative SBML model as a proof-of-principle for the methodology.
The context for the use cases is based on the Proctor and colleagues’ S.cerevisiae telomere model previously described in this thesis [39]. In this way the integration results for the use cases can be compared against an already-extant, curated model. One of the key proteins within this model, RAD9, was chosen as the basis for the two use cases. In the use cases, various types of information about this query protein are retrieved. These examples highlight how rule-based mediation works in a systems biology setting and provide insight into how such a system might be extended in the future to provide automated model annotation.
Each use case shows a different aspect of the integration process. Use Case 1 shows how an existing entity within an SBML model can be augmented, and Use Case 2 describes the addition of near neighbours to the selected model species. The use cases begin with the same initial conditions. The telomere ontology described in Section 4.1 was used as the core ontology for the sources described in Section 2. Three standard Semantic Web tools (XMLTab [31], SWRLTab and SQWRLQueryTab [40]) were then used to implement the integration step via SWRL mappings. The information retrieved from the core ontology allowed the annotation of an SBML model, including the provisional identification of information not previously present in the model. These use cases demonstrate the feasibility of the approach, the applicability of the technology and its utility in gaining new knowledge.
The following subsections describe two methods for enriching the telomere model. In Use Case 1, a single SBML species is annotated with information relevant to the gene RAD9. Adding information to existing SBML elements at an early stage aids model development and extension prior to publication or deposition. In Use Case 2, possible protein-protein interactions are retrieved involving the RAD9 protein. This approach results in the identification of model elements as well as a putative match for an enzyme that was not identified in the original curated model. These examples show how rule-based mediation works in a systems biology setting. These use cases also show that rule-based mediation successfully integrates information from multiple sources using existing tools, and that it would be useful to expand the implementation of this method to additional biological questions and automated model annotation.
Prior to performing the queries for each use case, all mapping rules from the source ontologies to the telomere ontology were run (BP_*, PSIMIF_*, and UP_*, as described in Section 3). These rules populated the telomere ontology with the instance data from the source ontologies. Next, the rules to further classify the new instance data within the telomere ontology (TUO_000*) were executed. Once the execution of the SWRL rules was complete, the telomere ontology can be queried with SQWRL. To get the final results, the queries and rules specific to the use cases (TUO_MFO_* and TUO_SQWRL_*) are executed.
Protégé 3.4 and the built-in Pellet 1.5.2 reasoner were used to reason over the entire set of integrated ontologies as well as the SBML model within MFO. When running on a dual-CPU laptop running Ubuntu 11.10 with 4 Gigabytes of memory, consistency checks for the set of integrated ontologies take approximately 10-15 seconds, while computing inferred types requires approximately 9-12 minutes. Reasoning over just MFO with the annotated model from the use case takes less than 10 seconds for both consistency checks and the computation of inferred types. Finally, the MFO software toolset was used to convert the newly-annotated model from OWL to SBML.
Once instance data is loaded into telomere ontology from all of the source ontologies via the rules described in Section 3, reasoning as well as the application of telomere ontology specific rules can be performed for the merging of semantically identical instances, logical inconsistencies and the inference of instances to their most specific location in the hierarchy. For each use case, a query term and a taxonomic restriction has been provided. The query results are mapped from the telomere ontology to either a new or existing SBML model in MFO.
5.1 Use Case 1: Augmentation of existing model species
In Use Case 1, the SBML species ’rad9’ is investigated. The only information provided by the modeller is the query term ’rad9’ and the taxonomic species S.cerevisiae. In contrast to a taxonomic species, an SBML species is any physical entity that can be part of an SBML reaction. There are a number of motivations for such a query: many models are initially created as skeletons, with all species and reactions present but without accompanying data; alternatively, modellers might be searching for annotation at an even earlier stage, and want to see what is available for just one particular species. By adding information to SBML components at an early stage, modellers will not only have a more complete model, but such extra information could also aid them in developing and extending the model prior to publication or deposition in a database such as BioModels [29]. This use case showcases the ability of the methodology to add basic information to the species such as cross-references and other annotations.
Therefore in Use Case 1, the telomere ontology was queried for information about the S.cerevisiae gene RAD9. This use case demonstrates the addition of basic information to the species such as cross-references, SBO annotations, compartment localisations and a recommended name. The query for proteins containing ’rad9’ as a name or synonym are stored within the telomere ontology as SWRL rules TUO_00001-2 (described initially in Section 4.2). Running the rules collects all matching telomere ontology Protein instances and stores them as instances of Rad9, a subclass of Protein.
The antecedent of TUO_00001 queries the telomere ontology synonym property within Protein for a string matching “rad9”, and the consequent of the rule fills Rad9 with any matched instances. TUO_00002 works in the same way, with recommendedName as the queried field. Running these SWRL rules places the following two instances under Rad9, each from a different data source:
cpath:CPATH-92332
upkb:protein_0
The next step is to discover equivalent instances elsewhere in the integrated schema and mark them as such. This is done by mimicking the behaviour of the OWL 2 construct owl:hasKey (see also Section 6). The end result of this procedure is that all instances with the same SGD accession will be marked as identical using the owl:sameAs construct. In future, this can happen automatically as the relation linking a telomere ontology Protein to its SGD accession number can be classed as the key for the Protein class in an owl:hasKey construct.
Until the owl:hasKey construct is used, those instances having the same SGD accession can be identified using a SQWRL query such as that shown below, and then manually adding the owl:sameAs assertions. A SQWRL is more appropriate, as SWRL rules modify the target ontology.
tuo:Protein(?someEntity) ∧
tuo:hasDatabaseReference(?someEntity, ?dbref ) ∧
tuo:databaseName(?dbref, ?name) ∧
swrlb:containsIgnoreCase(?name, “SGD” ) ∧
tuo:accession(?dbref, ?acc) ∧
swrlb:equal(?acc, “S000002625” ) →
sqwrl:selectDistinct(?someEntity)
There are already two instances of Rad9 as a result of TUO_00001-2. After running the SQWRL query above and viewing the results (upkb:protein_0 and psimif:interactor_59), the owl:sameAs construct can be applied between those two instances. The new instance, psimif:interactor_59, does not contain ‘rad9’ in its name or synonyms, but does contain a matching SGD accession. After inferring the placement of all individuals in the ontology using a reasoner, psimif:interactor_59 is inferred as an instance of Rad9, bringing the total number of Rad9 instances to three:
cpath:CPATH-92332
upkb:protein_0
psimif:interactor_59
The final missing owl:sameAs equivalences were then declared for these three instances, allowing the knowledge contained within each of them to be accessible as a single logical unit. The final step of this use case is to restrict the instances of Rad9 to the specified NCBI taxonomic identifier for S.cerevisiae. The SQWRL query TUO_SQWRL_00001 (see Section 4.2) performs this function.
The information contained within these three instances is then sent out, via MFO, to a new version of the telomere model. In order to export the relevant information to MFO, a number of mapping rules with telomere ontology classes in the antecedent and MFO classes in the consequent were created (see Section 3.4). To create MIRIAM cross-references and SBO terms, rules TUO_MFO_00001 through TUO_MFO_00004 were run. For instance, in TUO_MFO_00001, SBO terms and all UniProtKB cross-references for Rad9 were retrieved and reformatted according to MIRIAM guidelines, and added to the appropriate MFO Rad9 species.
The original curated telomere model contained a single reference to UniProtKB, which was confirmed by the information contained within the telomere ontology. In addition, an SBO term and a number of other MIRIAM annotations were exported into the model for the two RAD9 species in the model. As described in Section 3.4, Protein classes from the telomere ontology are mapped directly to species elements in SBML. In Use Case 1, mapping from the telomere ontology back to MFO links annotation from the relevant Rad9 instances back to MIRIAM annotations within the MFO Species class. The XML snippet below shows one of the Rad9 species after the results of this use case were exported:
<species metaid="_044468" id="Rad9A" name="Rad9" compartment="nucleus"
initialAmount="0" hasOnlySubstanceUnits="true" sboTerm="SBO:0000245">
<annotation>
<rdf:RDF>
<bqbiol:is>
<rdf:li rdf:resource="urn:miriam:uniprot:P14737"/>
<rdf:li rdf:resource="urn:miriam:intact:EBI-14788"/>
<rdf:li rdf:resource="urn:miriam:uniprot:Q04920"/>
<rdf:li rdf:resource="urn:miriam:pathwaycommons:92332"/>
<rdf:li rdf:resource="urn:miriam:sgd:S000002625"/>
<rdf:li rdf:resource="urn:miriam:intact:P14737"/>
</bqbiol:is>
</rdf:RDF>
</annotation>
</species>
All three data sources contributed to the set of information retrieved in Use Case 1. By successfully mapping data from the source ontologies into the core telomere ontology, information was retrieved regarding proteins with ’rad9’ as one of their names. This information was then added to an SBML file by mapping from the telomere ontology back to MFO.
The use of the owl:sameAs construct for identifying equivalent instances is helpful, but caused some problems for this use case. For instance, a bug the SQWRL implementation used in this work prevented the use of any instances marked with owl:sameAs axioms, and these axioms were removed prior to running the queries. In addition, there is the potential for false positives in defining two instances as identical using owl:sameAs. For instance, BioPAX has multiple types of cross-references including unificationXref (for cross-references linking to semantically identical entries in the other data resources) and relationshipXref, which implies a less rigorous link. If the wrong type of key is created (e.g. using relationshipXref rather than unificationXref), or an imprecise rule is used, then two instances could be incorrectly marked as identical. Mapping rules must therefore be created carefully, or they could lead to false positives with respect to related, but not identical, instances.
5.2 Use Case 2: Augmentation by addition of near neighbours
Use Case 2 shows how information about possible protein-protein interactions involving RAD9 can be retrieved from the telomere ontology, confirming interactions already present in the curated model as well as discovering novel interactions. The retrieved information results in the proposal of new model elements as well as the putative identification of the enzyme responsible for activation of RAD9. The results of Use Case 1 are used as part of the input in this use case. TUO_SQWRL_00004 is the SQWRL query used to display, but not store, all interactions involving Rad9 (see also Section 4.2).
In TUO_SQWRL_00004, the antecedent of the SQWRL query identifies interactions involving Rad9 instances via the Participant class. The consequent displays the name of the instance, and the process in which it is involved. The use of SQWRL here rather than SWRL allows viewing of the more than 270 returned interactions. These results are not saved in the asserted ontology as there are too many, some of which are not meaningful for the model under study. If necessary, such unwanted results could be excluded with a small modification to the query or to the output mapping rules for MFO. Here, the interactions retrieved in TUO_SQWRL_00004 were filtered according to those species already present in the model, many of which did not have any prior involvement with RAD9. Before such relevant interactions can be found, the identity of the proteins involved must be determined. For example, just as Rad9 instances were identified in Use Case 1, so should all other proteins of interest be identified. While these rules are currently manually generated, they could easily be automatically created and run on-the-fly. TUO_00001-11 are the SWRL rules which populates the class for these proteins of interest (see Section 4.2).
The combined results from queries TUO_00001-11 result in the appropriate proteins for this model being identified and classified. Once this is complete, specific interactions of interest can also be identified. Below are the OWL axioms on the telomere ontology defined class Rad9Rad53Interaction. This is one of four such defined classes which filter the Rad9 interactions. When reasoning over instance data is performed, the appropriate PhysicalProcess instances which meet the criteria set out in these necessary and sufficient axioms are inferred to be children of Rad9Rad53Interaction.
Class: Rad9Rad53Interaction
EquivalentTo:
(hasParticipant some (playedBy some Rad53))
and (hasParticipant some (playedBy some Rad9))
SubClassOf:
Rad9Interaction
Table 2 shows a summary of the inference results from the defined interaction classes
Rad9Rad53Interaction, Rad9Chk1Interaction, Rad9Mec1Interaction and Rad9Rad17Interaction. The results may also be obtained by running TUO_00012-15 (see Section 4.2), or by loading the asserted ontology and re-running the reasoning. There are a total of four interactions involving RAD9 present in the curated model. Two of these, with RAD53 and CHK1, were confirmed in this use case. The RAD53 interaction was found in both BioGRID and in Pathway Commons, while the CHK1 interaction was only in Pathway Commons.
| Discovered Interactor with RAD9 (P14737) |
Proctor et al. |
BioGRID |
Pathway Commons |
| Serine/threonine-protein kinase RAD53 (P22216) |
yes |
yes |
yes |
| Serine/threonine-protein kinase CHK1 (P38147) |
yes |
|
yes |
| Serine/threonine-protein kinase MEC1 (P38111) |
|
yes |
|
| DNA damage checkpoint control RAD17 (P48581) |
|
yes |
|
| Rad9Kin (*) |
yes |
|
|
| ExoX (*) |
yes |
|
|
Table 2: Summary of interactions retrieved for Use Case 2 against the telomere ontology. All discovered interactions already present in the curated model (RAD53 and CHK1) are shown, together with example interactions from the curated model that were not discovered (with Rad9Kin and ExoX) and discovered interactions that were not present in the model (with MEC1 and RAD17). SBML species shown with an asterisk (*) are those which are placeholder species, and therefore cannot have a match to a real protein. Some interactions are false positives inherent in the data source, while others are out of scope of the modelling domain of interest and should not be not included.
The other two RAD9 interactions are placeholder species, created by the modeller to describe an unknown protein which could not be matched further. In addition to confirming the RAD53 and CHK1 interactions with RAD9, one of those placeholder species, marked as ’Rad9Kin’ in the model, was provisionally identified as the protein MEC1 using data originating in BioGRID. In the curated model, the ’rad9Kin’ species does not have a UniProtKB primary accession, as the model author did not know which protein activated RAD9. However, MEC1 is shown in the telomere ontology as interacting with RAD9 and is present elsewhere in the curated model reactions. Further, UniProtKB reports it as a kinase which phosphorylates RAD9. From this information, the model author now believes that MEC1 could be the correct protein to use as the activator of RAD9 [41]. RAD17 was added to Table 2 as another example of a protein present in the curated model, but not as an interacting partner of RAD9. This interaction may have been unknown to the modeller, and it may be that the curated model could be improved by the addition of a RAD9/RAD17 reaction.
After the information was retrieved within the telomere ontology, SWRL rules TUO_MFO_00005-12 were run to export the information from the telomere ontology into MFO and, ultimately, into the original SBML model. The XML snippet below shows the new SBML species added to the model as part of the new reaction with MEC1:
<species metaid="_3a9a2e27" id="interactor_24_id" name="YBR136W"
sboTerm="SBO:0000245">
<annotation>
<rdf:RDF>
<rdf:Description rdf:about="#_3a9a2e27">
<bqbiol:is>
<rdf:Bag>
<rdf:li rdf:resource="urn:miriam:sgd:S000000340"/>
<rdf:li rdf:resource="urn:miriam:uniprot:P38111"/>
</rdf:Bag>
</bqbiol:is>
</rdf:Description>
</rdf:RDF>
</annotation>
</species>
Here, the id is based on the name of the telomere ontology Mec1 instance, the name is that instance’s recommendedName or synonym, and the MIRIAM annotations are drawn from the list of DatabaseReference instances. MIRIAM annotations, names and SBO terms were added to the SBML species referenced in the use cases, and two example reactions were added: RAD9 with RAD17, and RAD9 with MEC1. The former was chosen as an example of how a ProteinComplexFormation reaction is mapped back to MFO, and the latter was chosen as an example of the mapping of a simpler interaction. Further, as MEC1 is a candidate for the value of the ’Rad9Kin’ species in the curated model, creating a skeleton reaction in the annotated version of the curated model is the first step in updating the model. An excerpt of the RAD9/RAD17 reaction in SBML is included below. Further work by the modeller can flesh out the base reaction, species and annotation created by the integration method to include parameters, initial values and rate constants.
<reaction id="interaction_160_id">
<listOfReactants>
<speciesReference species="interactor_59_id"/>
<speciesReference species="interactor_8_id"/>
</listOfReactants>
<listOfProducts>
<speciesReference species="product_of_rad9rad17interaction"/>
</listOfProducts>
</reaction>
[...]
<species metaid="_720ffc30" id="interactor_8_id" name="YOR368W"
sboTerm="SBO:0000245">
<annotation>
<rdf:RDF>
<rdf:Description rdf:about="#_720ffc30">
<bqbiol:is>
<rdf:Bag>
<rdf:li rdf:resource="urn:miriam:sgd:S000005895"/>
<rdf:li rdf:resource="urn:miriam:uniprot:P48581"/>
</rdf:Bag>
</bqbiol:is>
</rdf:Description>
</rdf:RDF>
</annotation>
</species>
<species id="product_of_rad9rad17interaction" name="Rad9Rad17Complex"/>
[...]
For Use Case 2, while BioGRID contained the most interactions, it was UniProtKB and Pathway Commons which together contributed the majority of the other types of information. Curated RAD9 reactions with RAD53 and CHK1 were recovered, while of the many other reactions, one (with RAD17) was with an existing species in the model and another (with MEC1) putatively identified a placeholder species in that model.
Within the telomere ontology, the product of a protein complex formation must be a protein complex as described in the ProteinComplexFormation class. Specifically the RAD9/RAD53 and RAD9/RAD17 reactions are part of a BioGRID ‘Reconstituted Complex’. This reaction type can be presented for RAD9/RAD53 as  . While not identical to the curated reaction, which shows activation of RAD53 by RAD9 (
. While not identical to the curated reaction, which shows activation of RAD53 by RAD9 (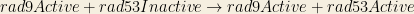 ), the proposed interaction is potentially informative.
), the proposed interaction is potentially informative.
Use Case 2 addresses a modeller’s need to learn of possible new reactions involving the query species retrieved from Use Case 1. In this scenario, the modeller is only interested in protein-protein interactions. The query for this use case would be beneficial during model creation or extension of an already-extant model.
While BioGRID contained the most interactions for the second use case, it was UniProtKB and Pathway Commons which together contributed the majority of the information beyond those interactions. Combining data sources in a semantically-meaningful, rule-based mediation methodology is a useful way of retrieving useful information for SBML model annotation.
6 Future directions
6.1 Data provenance
There are limitations in describing data provenance using the SBML standard notation. When new MIRIAM annotations are added to an SBML model, there is no way of stating what procedure added the annotations, or the date they were added. At a minimum, the addition of the software type, version number and date on which the information was added should be included. At the moment, SBML is unable to make statements about annotations. The proposed annotation package for SBML Level 3 will address this issue, among others [42]. Until this issue is resolved, no provenance information is included in the newly-annotated models.
6.2 Mapping and the creation of syntactic ontologies
When mapping the telomere ontology to MFO, a number of limitations were encountered with the technology used. For instance, in order to map a telomere ontology PhysicalProcess such as ProteinComplexFormation correctly, a number of new instances were created when the rule was run (see rule in Section 3.4). Then, when multiple reactions make use of a single protein instance (such as, in these use cases, any Rad9 instance), a new MFO identifier, or SId, is created. The ultimate result is that more than one SId was created for the same protein instance, which if left alone would result in an inconsistent ontology, as only one SId is allowed per MFO Species. Also, because SWRL rules do not add information to a data property slot unless that slot is already empty, running a rule which adds a telomere ontology recommendedName value to an MFO species will not work if a similar rule using the synonym value has previously been run. These are all limitations that can be ameliorated through more sophisticated usages of OWL and SWRL tools. One such tool already used within the MFO project is the OWL API [43], which allows finer control over ontologies and their mapping and reasoning. Greater use of the SWRLAPI [32] might also address these limitations.
While extensive use of pre-existing applications, plugins, and libraries has been made, in the very near future the limit of some of these technologies could be reached. Particularly, improvements will need to be made to the way the XML is converted into OWL. Further, if this methodology is to be used in a larger-scale semantic data integration project, the correspondingly large number of instances required will most likely necessitate the use of a database back-end for the ontologies. Some technologies are available for storing instances from ontologies within a database schema such as that followed by Sahoo and colleagues [8], the Protégé database back-end (Note: http://protegewiki.stanford.edu/wiki/Working_with_the_Database_Backend_in_OWL) or DBOWL [44].
6.3 OWL 2
The use cases presented in this chapter, while performed with existing tools, required manual intervention. Improvements can be made to the implementation of this methodology through the use of additional OWL 2 features. In order to assert equivalences between instance data from the various data sources, the owl:sameAs construct was manually applied. Tool and reasoning support for OWL 2, now a W3C Recommendation [45], is growing, and with OWL 2, some advances such as the owl:hasKey [46] construct allows more automation in this area. These keys, or inverse functional datatype properties, allow the definition of equivalence rules based on properties such as database accessions, which in the current system was achieved by manual mapping. For example, by applying the owl:hasKey axiom to a data property linking to UniProtKB accession numbers, any instances containing matching UniProtKB accessions will be marked as identical.
6.4 Classes versus instance modelling for biological entities
In ontologies such as BioPAX Level 3, a specific protein is modelled as an instance of a class rather than as a class in its own right. For instance, RAD9 would be an instance of the BioPAX Protein class. Conversely in the telomere ontology, every protein—including Rad9—has been given its own class. This allows for expressive statements to be made about the protein and about particular interactions the protein is involved in. While this type of mapping allows greater expressivity, there are challenges to the broader usefulness of such mapping. As Demir has highlighted (Note: https://groups.google.com/forum/?hl=en#!topic/biopax2model/01XfxFfLxhA), the these hurdles are social as well as technological in nature.
Technologically, the use of expressive classes for specific biological entities such as Rad9 makes the information difficult for a “traditional” bioinformatician to use, as reasoners must be applied to get the most out of such ontologies. Reasoners are difficult for people unused to them, as their purpose is often unclear to new users and they tend to have esoteric error messages. From a social perspective, allowing users to create an unlimited number and type of entities such as Rad9 can lead to a lack of standard methods of creating and using those entities.
However, these challenges are all associated with the creation of an ontology which is expressly developed as a community, or shared, ontology. Core ontologies in rule-based mediation may be used by multiple groups, but are primarily intended to address a specific and possibly short-term biological question, and as such do not need to fulfil such a wide remit. While the initial development of the source and core ontologies would require an ontologist, the populating of the resulting knowledgebase within the core ontology is intended to be accessible via a Web application currently under development and, in future, the querying of that knowledgebase via Saint (Chapter 3). Reasoners are an intrinsic part of rule-based mediation, and the number of ontologists building the core ontology would normally be small. Therefore, as the issues raised by Demir are valid for large-scale ontologies intended for public use, they do not apply to the primary role of rule-based mediation.
In the use cases described in this chapter, instance-to-instance SWRL mappings assign membership of instances from source ontologies to classes from the core ontology. For example, the rad9A instance from the MFO version of the telomere model becomes an instance of the Rad9 class in the telomere ontology. It would be useful to be able to use mappings to automatically create core ontology classes such as Rad9. This instance-to-class mapping is theoretically possible, but no programmatically-accessible rule language has this feature. Such rules could instead be hard-coded using a library such as the OWL API. This would allow the automated population of a core ontology with all of the necessary classes for a particular pathway or systems biology model of interest.
7 Discussion
In rule-based mediation, a set of source ontologies are individually linked to a core ontology, which describes the domain of interest. Similar or even overlapping data sources and formats can be handled, as each format will have its own source ontology. Although some researchers question the conversion from a closed-world syntax such as XML to a open-world language such as OWL (Note: https://groups.google.com/forum/?hl=en#!topic/biopax2model/01XfxFfLxhA), the use of syntactic ontologies allows the separation of the closed-world source syntax from the biological domain modelling which takes place in the core ontology. In essence, the OWL in a syntactic ontology is “abused” in order to allow the conversion of the underlying source data to the more expressive model present in the core ontology. Unlike previous integration methods where one ontology type is created as a view of the other, rule-based mediation allows both the straightforward addition of new source ontologies as well as the maintenance of the core ontology as an independent entity. Using a core ontology allows new data sources and data formats to be added without changing the way the core model of the biological domain is structured.
Rule-based mediation was shown to be a valid methodology for two use cases relevant to the biological modelling community. Two syntactic ontologies were created to represent PSI-MIF and UniProtKB XML entries in OWL format. BioPAX has also been adopted as one of the source ontologies, showing how pre-existing ontologies can successfully be used as a source ontology without modification. The telomere ontology, which describes the research domain surrounding the use cases, can be reasoned over and is able to store information mapped from multiple source ontologies. The source ontologies and telomere ontology are available for download, pre-filled with the use case instances. In future, a Web application will allow the user to query the source databases and retrieve a tailor-made dataset which is then automatically mapped to the telomere ontology. A semantic, rule-based data integration methodology has been created, and proof-of-principle for that methodology in the context of SBML model annotation has been shown.
use it, or would there be no need for instances at all? We will need instances of 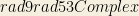 to show that a particular data source does represent this reaction.)
to show that a particular data source does represent this reaction.)
Bibliography
- [1]
- Peter Li, Tom Oinn, Stian Soiland, and Douglas B. Kell. Automated manipulation of systems biology models using libSBML within Taverna workflows. Bioinformatics (Oxford, England), 24(2):287–289, January 2008.
- [2]
- Falko Krause, Jannis Uhlendorf, Timo Lubitz, Marvin Schulz, Edda Klipp, and Wolfram Liebermeister. Annotation and merging of SBML models with semanticSBML. Bioinformatics, 26(3):421–422, February 2010.
- [3]
- Neil Swainston and Pedro Mendes. libAnnotationSBML: a library for exploiting SBML annotations. Bioinformatics, 25(17):2292–2293, September 2009.
- [4]
- M. L. Blinov, O. Ruebenacker, and I. I. Moraru. Complexity and modularity of intracellular networks: a systematic approach for modelling and simulation. IET systems biology, 2(5):363–368, September 2008.
- [5]
- Allyson L. Lister, Matthew Pocock, Morgan Taschuk, and Anil Wipat. Saint: a lightweight integration environment for model annotation. Bioinformatics, 25(22):3026–3027, November 2009.
- [6]
- Natalya F. Noy and Mark A. Musen. The PROMPT suite: interactive tools for ontology merging and mapping. Int. J. Hum.-Comput. Stud., 59(6):983–1024, December 2003.
- [7]
- Robert Hoehndorf, Michel Dumontier, Anika Oellrich, Dietrich Rebholz-Schuhmann, Paul N. Schofield, and Georgios V. Gkoutos. Interoperability between Biomedical Ontologies through Relation Expansion, Upper-Level Ontologies and Automatic Reasoning. PLoS ONE, 6(7):e22006+, July 2011.
- [8]
- Satya S. Sahoo, Olivier Bodenreider, Joni L. Rutter, Karen J. Skinner, and Amit P. Sheth. An ontology-driven semantic mashup of gene and biological pathway information: application to the domain of nicotine dependence. Journal of biomedical informatics, 41(5):752–765, October 2008.
- [9]
- O. Ruebenacker, I. I. Moraru, J. C. Schaff, and M. L. Blinov. Integrating BioPAX pathway knowledge with SBML models. IET Systems Biology, 3(5):317–328, 2009.
- [10]
- Li Xu and David W. Embley. Combining the Best of Global-as-View and Local-as-View for Data Integration. In Anatoly E. Doroshenko, Terry A. Halpin, Stephen W. Liddle, Heinrich C. Mayr, Anatoly E. Doroshenko, Terry A. Halpin, Stephen W. Liddle, and Heinrich C. Mayr, editors, ISTA, volume 48 of LNI, pages 123–136. GI, 2004.
- [11]
- Alan Ruttenberg, Jonathan A. Rees, Matthias Samwald, and M. Scott Marshall. Life sciences on the Semantic Web: the Neurocommons and beyond. Briefings in bioinformatics, 10(2):193–204, March 2009.
- [12]
- Michael Bada, Kevin Livingston, and Lawrence Hunter. An Ontological Representation of Biomedical Data Sources and Records. Ontogenesis, June 2011.
- [13]
- Marvin Schulz, Falko Krause, Nicolas Le Novere, Edda Klipp, and Wolfram Liebermeister. Retrieval, alignment, and clustering of computational models based on semantic annotations. Molecular Systems Biology, 7(1), July 2011.
- [14]
- Robert Hoehndorf, Michel Dumontier, John Gennari, Sarala Wimalaratne, Bernard de Bono, Daniel Cook, and Georgios Gkoutos. Integrating systems biology models and biomedical ontologies. BMC Systems Biology, 5(1):124+, August 2011.
- [15]
- R. Stevens, P. Baker, S. Bechhofer, G. Ng, A. Jacoby, N. W. Paton, C. A. Goble, and A. Brass. TAMBIS: transparent access to multiple bioinformatics information sources. Bioinformatics (Oxford, England), 16(2):184–185, February 2000.
- [16]
- Marie C. Rousset and Chantal Reynaud. Knowledge representation for information integration. Inf. Syst., 29(1):3–22, 2004.
- [17]
- Ian Horrocks, Peter F. Patel-Schneider, Harold Boley, Said Tabet, Benjamin Grosof, and Mike Dean. SWRL: A Semantic Web Rule Language Combining OWL and RuleML. http://www.w3.org/Submission/SWRL/, May 2004.
- [18]
- Bijan Parsia. Understanding SWRL (Part 1), August 2007.
- [19]
- Pascal Hitzler. OWL and Rules. In OWLED 2011, June 2011.
- [20]
- Markus Krötzsch, Sebastian Rudolph, and Pascal Hitzler. Description Logic Rules. In Proceeding of the 2008 conference on ECAI 2008: 18th European Conference on Artificial Intelligence, pages 80–84, Amsterdam, The Netherlands, The Netherlands, 2008. IOS Press.
- [21]
- Martin O’Connor, Holger Knublauch, Samson Tu, Benjamin Grosof, Mike Dean, William Grosso, and Mark Musen. Supporting Rule System Interoperability on the Semantic Web with SWRL. pages 974–986. 2005.
- [22]
- M. J. O’Connor and A. K. Das. SQWRL: a Query Language for OWL. In OWL: Experiences and Directions (OWLED), Fifth International Workshop, 2009.
- [23]
- Aitken Stuart, Korf Roman, Webber Bonnie, and Bard Jonathan. COBrA: a bio-ontology editor. Bioinformatics, 21(6):825–826, March 2005.
- [24]
- S. M. Falconer, N. F. Noy, and M. A. Storey. Ontology Mapping – A User Survey. In The Second International Workshop on Ontology Matching at ISWC 07 + ASWC 07, 2007.
- [25]
- Joanne S. Luciano and Robert D. Stevens. e-Science and biological pathway semantics. BMC bioinformatics, 8 Suppl 3(Suppl 3):S3+, 2007.
- [26]
- Chris Stark, Bobby-Joe Breitkreutz, Teresa Reguly, Lorrie Boucher, Ashton Breitkreutz, and Mike Tyers. BioGRID: a general repository for interaction datasets. Nucleic Acids Research, 34(suppl 1):D535–D539, January 2006.
- [27]
- Henning Hermjakob, Luisa Montecchi-Palazzi, Gary Bader, Jérôme Wojcik, Lukasz Salwinski, Arnaud Ceol, Susan Moore, Sandra Orchard, Ugis Sarkans, Christian von Mering, Bernd Roechert, Sylvain Poux, Eva Jung, Henning Mersch, Paul Kersey, Michael Lappe, Yixue Li, Rong Zeng, Debashis Rana, Macha Nikolski, Holger Husi, Christine Brun, K. Shanker, Seth G. Grant, Chris Sander, Peer Bork, Weimin Zhu, Akhilesh Pandey, Alvis Brazma, Bernard Jacq, Marc Vidal, David Sherman, Pierre Legrain, Gianni Cesareni, Ioannis Xenarios, David Eisenberg, Boris Steipe, Chris Hogue, and Rolf Apweiler. The HUPO PSI’s molecular interaction format–a community standard for the representation of protein interaction data. Nature biotechnology, 22(2):177–183, February 2004.
- [28]
- Ethan G. Cerami, Benjamin E. Gross, Emek Demir, Igor Rodchenkov, Özgün Babur, Nadia Anwar, Nikolaus Schultz, Gary D. Bader, and Chris Sander. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Research, 39(suppl 1):D685–D690, January 2011.
- [29]
- Chen Li, Marco Donizelli, Nicolas Rodriguez, Harish Dharuri, Lukas Endler, Vijayalakshmi Chelliah, Lu Li, Enuo He, Arnaud Henry, Melanie Stefan, Jacky Snoep, Michael Hucka, Nicolas Le Novere, and Camille Laibe. BioModels Database: An enhanced, curated and annotated resource for published quantitative kinetic models. BMC Systems Biology, 4(1):92+, June 2010.
- [30]
- The UniProt Consortium. The Universal Protein Resource (UniProt). Nucl. Acids Res., 36(suppl_1):D190–195, January 2008.
- [31]
- Michael Sintek. XML Tab. http://protegewiki.stanford.edu/wiki/XML_Tab, June 2008.
- [32]
- M. J. O’Connor, R. D. Shankar, S. W. Tu, C. I. Ny, and A. K. Dasulas. Developing a Web-Based Application using OWL and SWRL. In AAAI Spring Symposium, 2008.
- [33]
- Christoph Lange. Krextor — An Extensible XML->RDF Extraction Framework. In Chris Bizer, Sören Auer, and Gunnar A. Grimnes, editors, 5th Workshop on Scripting and Development for the Semantic Web, Colocated with ESWC 2009, May 2009.
- [34]
- A. L. Lister, M. Pocock, and A. Wipat. Integration of constraints documented in SBML, SBO, and the SBML Manual facilitates validation of biological models. Journal of Integrative Bioinformatics, 4(3):80+, 2007.
- [35]
- Michael Hucka. SBML Groups Proposal (2009-09). http://sbml.org/Community/Wiki/SBML_Level_3_Proposals/Groups_Proposal_%282009-09%29, September 2009.
- [36]
- Holger Wache, T. Vögele, Ubbo Visser, H. Stuckenschmidt, G. Schuster, H. Neumann, and S. Hübner. Ontology-based integration of information — a survey of existing approaches. In H. Stuckenschmidt, editor, Proceedings of the IJCAI’01 Workshop on Ontologies and Information Sharing, Seattle, Washington, USA, Aug 4-5, pages 108–117, 2001.
- [37]
- Jinguang Gu, Baowen Xu, and Xinmeng Chen. An XML query rewriting mechanism with multiple ontologies integration based on complex semantic mapping. Information Fusion, 9(4):512–522, October 2008.
- [38]
- J. S. Luciano. PAX of mind for pathway researchers. Drug discovery today, 10(13):937–942, July 2005.
- [39]
- C. J. Proctor, D. A. Lydall, R. J. Boys, C. S. Gillespie, D. P. Shanley, D. J. Wilkinson, and T. B. L. Kirkwood. Modelling the checkpoint response to telomere uncapping in budding yeast. Journal of The Royal Society Interface, 4(12):73–90, February 2007.
- [40]
- Martin O’Connor. SQWRLQueryTab. http://protege.cim3.net/cgi-bin/wiki.pl?SQWRLQueryTab, February 2010.
- [41]
- C. J. Proctor. Personal Communication, 2009.
- [42]
- Dagmar Waltemath, Neil Swainston, Allyson Lister, Frank Bergmann, Ron Henkel, Stefan Hoops, Michael Hucka, Nick Juty, Sarah Keating, Christian Knuepfer, Falko Krause, Camille Laibe, Wolfram Liebermeister, Catherine Lloyd, Goksel Misirli, Marvin Schulz, Morgan Taschuk, and Nicolas Le Novère. SBML Level 3 Package Proposal: Annotation. Nature Precedings, (713), January 2011.
- [43]
- Matthew Horridge, Sean Bechhofer, and Olaf Noppens. Igniting the OWL 1.1 Touch Paper: The OWL API. In Proceedings of OWLEd 2007: Third International Workshop on OWL Experiences and Directions, 2007.
- [44]
- Mar
 a del Mar d. e. l. . M. Roldán-Garca, Ismael Navas-Delgado, Amine Kerzazi, Othmane Chniber, Joaqun Molina-Castro, and José F. Aldana-Montes. KA-SB: from data integration to large scale reasoning. BMC bioinformatics, 10 Suppl 10(Suppl 10):S5+, 2009.
a del Mar d. e. l. . M. Roldán-Garca, Ismael Navas-Delgado, Amine Kerzazi, Othmane Chniber, Joaqun Molina-Castro, and José F. Aldana-Montes. KA-SB: from data integration to large scale reasoning. BMC bioinformatics, 10 Suppl 10(Suppl 10):S5+, 2009.
- [45]
- The W3C OWL Working Group. OWL 2 Web Ontology Language Document Overview. http://www.w3.org/TR/owl2-overview/, October 2009.
- [46]
- Christine Golbreich and Evan K. Wallace. hasKey, in OWL 2 Web Ontology Language: New Features and Rationale. http://www.w3.org/TR/owl2-new-features/#F9:a_Keys, October 2009.

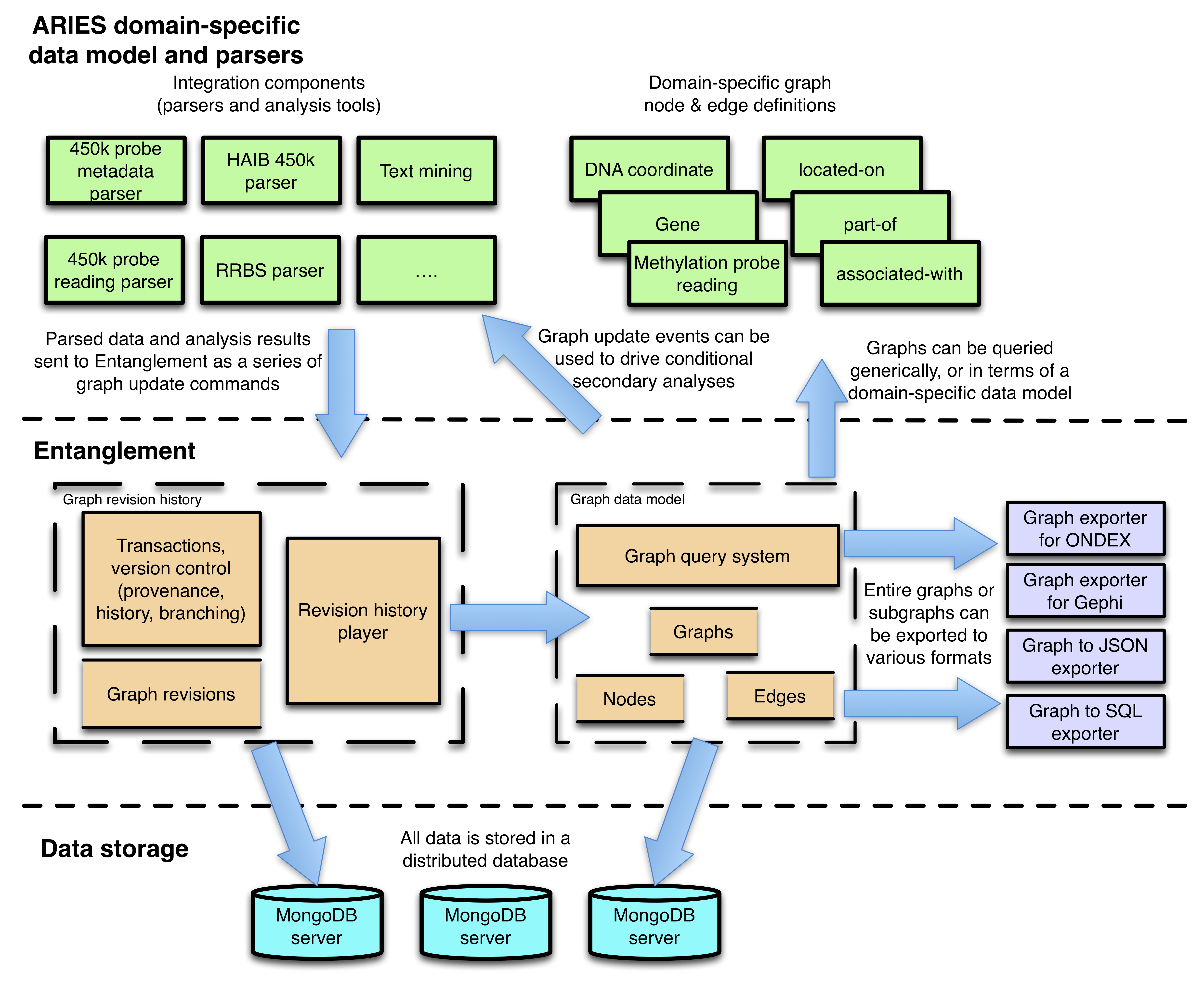




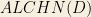






















 factor” it was unclear which
factor” it was unclear which 

















{kind=link}
{kind=link}
You must be logged in to post a comment.